当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Investigating Cryptic Binding Sites by Molecular Dynamics Simulations.
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2020-03-05 , DOI: 10.1021/acs.accounts.9b00613 Antonija Kuzmanic 1 , Gregory R Bowman 2 , Jordi Juarez-Jimenez 3 , Julien Michel 3 , Francesco L Gervasio 1, 4
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2020-03-05 , DOI: 10.1021/acs.accounts.9b00613 Antonija Kuzmanic 1 , Gregory R Bowman 2 , Jordi Juarez-Jimenez 3 , Julien Michel 3 , Francesco L Gervasio 1, 4
Affiliation
|
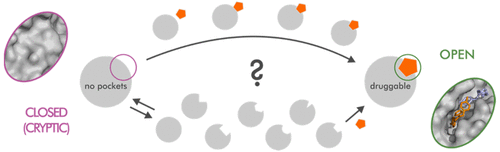
This Account highlights recent advances and discusses major challenges in investigations of cryptic (hidden) binding sites by molecular simulations. Cryptic binding sites are not visible in protein targets crystallized without a ligand and only become visible crystallographically upon binding events. These sites have been shown to be druggable and might provide a rare opportunity to target difficult proteins. However, due to their hidden nature, they are difficult to find through experimental screening. Computational methods based on atomistic molecular simulations remain one of the best approaches to identify and characterize cryptic binding sites. However, not all methods are equally efficient. Some are more apt at quickly probing protein dynamics but do not provide thermodynamic or druggability information, while others that are able to provide such data are demanding in terms of time and resources. Here, we review the recent contributions of mixed-solvent simulations, metadynamics, Markov state models, and other enhanced sampling methods to the field of cryptic site identification and characterization. We discuss how these methods were able to provide precious information on the nature of the site opening mechanisms, to predict previously unknown sites which were used to design new ligands, and to compute the free energy landscapes and kinetics associated with the opening of the sites and the binding of the ligands. We highlight the potential and the importance of such predictions in drug discovery, especially for difficult ("undruggable") targets. We also discuss the major challenges in the field and their possible solutions.
中文翻译:

通过分子动力学模拟研究隐蔽结合位点。
该报告重点介绍了最新进展,并讨论了通过分子模拟研究隐性(隐藏)结合位点的主要挑战。隐性结合位点在没有配体的情况下结晶的蛋白质靶中不可见,并且仅在结合事件发生时在晶体学上可见。这些位点已被证明是可药物治疗的,可能为难于靶向的蛋白质提供了难得的机会。但是,由于其隐藏的性质,很难通过实验筛选找到它们。基于原子分子模拟的计算方法仍然是识别和表征隐秘结合位点的最佳方法之一。但是,并非所有方法都同样有效。有些更适合快速探测蛋白质动力学,但不提供热力学或药物作用信息,而其他能够提供此类数据的人则在时间和资源上都要求很高。在这里,我们回顾了混合溶剂模拟,元动力学,马尔可夫状态模型以及其他增强的采样方法在隐蔽站点识别和表征领域的最新贡献。我们讨论了这些方法如何能够提供有关位点开放机制性质的宝贵信息,如何预测以前未知的位点,这些位点用于设计新的配体,以及如何计算与位点开放相关的自由能态势和动力学。配体的结合。我们强调了这种预测在药物发现中的潜力和重要性,尤其是对于困难的(“不可药物”)目标。我们还将讨论该领域的主要挑战及其可能的解决方案。
更新日期:2020-03-05
中文翻译:
通过分子动力学模拟研究隐蔽结合位点。
该报告重点介绍了最新进展,并讨论了通过分子模拟研究隐性(隐藏)结合位点的主要挑战。隐性结合位点在没有配体的情况下结晶的蛋白质靶中不可见,并且仅在结合事件发生时在晶体学上可见。这些位点已被证明是可药物治疗的,可能为难于靶向的蛋白质提供了难得的机会。但是,由于其隐藏的性质,很难通过实验筛选找到它们。基于原子分子模拟的计算方法仍然是识别和表征隐秘结合位点的最佳方法之一。但是,并非所有方法都同样有效。有些更适合快速探测蛋白质动力学,但不提供热力学或药物作用信息,而其他能够提供此类数据的人则在时间和资源上都要求很高。在这里,我们回顾了混合溶剂模拟,元动力学,马尔可夫状态模型以及其他增强的采样方法在隐蔽站点识别和表征领域的最新贡献。我们讨论了这些方法如何能够提供有关位点开放机制性质的宝贵信息,如何预测以前未知的位点,这些位点用于设计新的配体,以及如何计算与位点开放相关的自由能态势和动力学。配体的结合。我们强调了这种预测在药物发现中的潜力和重要性,尤其是对于困难的(“不可药物”)目标。我们还将讨论该领域的主要挑战及其可能的解决方案。
























 京公网安备 11010802027423号
京公网安备 11010802027423号