当前位置:
X-MOL 学术
›
ACS Pharmacol. Transl. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Mapping the Molecular Surface of the Analgesic NaV1.7-Selective Peptide Pn3a Reveals Residues Essential for Membrane and Channel Interactions.
ACS Pharmacology & Translational Science Pub Date : 2020-02-19 , DOI: 10.1021/acsptsci.0c00002 Alexander Mueller 1 , Zoltan Dekan 1 , Quentin Kaas 1 , Akello J Agwa 1 , Hana Starobova 1 , Paul F Alewood 1 , Christina I Schroeder 1 , Mehdi Mobli 2 , Jennifer R Deuis 1 , Irina Vetter 1, 3
ACS Pharmacology & Translational Science Pub Date : 2020-02-19 , DOI: 10.1021/acsptsci.0c00002 Alexander Mueller 1 , Zoltan Dekan 1 , Quentin Kaas 1 , Akello J Agwa 1 , Hana Starobova 1 , Paul F Alewood 1 , Christina I Schroeder 1 , Mehdi Mobli 2 , Jennifer R Deuis 1 , Irina Vetter 1, 3
Affiliation
|
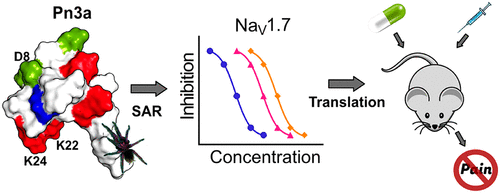
Compelling human genetic studies have identified the voltage-gated sodium channel NaV1.7 as a promising therapeutic target for the treatment of pain. The analgesic spider-venom-derived peptide μ-theraphotoxin-Pn3a is an exceptionally potent and selective inhibitor of NaV1.7; however, little is known about the structure–activity relationships or channel interactions that define this activity. We rationally designed 17 Pn3a analogues and determined their activity at hNaV1.7 using patch–clamp electrophysiology. The positively charged amino acids K22 and K24 were identified as crucial for Pn3a activity, with molecular modeling identifying interactions of these residues with the S3–S4 loop of domain II of hNaV1.7. Removal of hydrophobic residues Y4, Y27, and W30 led to a loss of potency (>250-fold), while replacement of negatively charged D1 and D8 residues with a positively charged lysine led to increased potencies (>13-fold), likely through alterations in membrane lipid interactions. Mutating D8 to an asparagine led to the greatest improvement in Pn3a potency at NaV1.7 (20-fold), while maintaining >100-fold selectivity over the major off-targets NaV1.4, NaV1.5, and NaV1.6. The Pn3a[D8N] mutant retained analgesic activity in vivo, significantly attenuating mechanical allodynia in a clinically relevant mouse model of postsurgical pain at doses 3-fold lower than those with wild-type Pn3a, without causing motor-adverse effects. Results from this study will facilitate future rational design of potent and selective peptidic NaV1.7 inhibitors for the development of more efficacious and safer analgesics as well as to further investigate the involvement of NaV1.7 in pain.
中文翻译:

绘制止痛药NaV1.7选择性肽Pn3a的分子表面图谱揭示了膜和通道相互作用必不可少的残基。
令人信服的人类遗传研究已经确定了电压门控钠通道Na V 1.7是治疗疼痛的有希望的治疗靶标。镇痛药源自蜘蛛毒液的肽μ-theraphotoxin-Pn3a是Na V 1.7的一种特别有效和选择性的抑制剂。但是,对于定义这种活动的结构-活动关系或渠道相互作用知之甚少。我们合理设计了17种Pn3a类似物,并使用膜片钳电生理学测定了它们在hNa V 1.7时的活性。带有正电荷的氨基酸K22和K24被确定对Pn3a活性至关重要,分子模型确定了这些残基与hNa V结构域II的S3–S4环之间的相互作用1.7。去除疏水残基Y4,Y27和W30会导致效价损失(> 250倍),而用带正电的赖氨酸替代带负电的D1和D8残基会导致效价增加(> 13倍),可能是通过膜脂质相互作用的改变。将D8突变为天冬酰胺可在Na V 1.7(20倍)时最大程度地提高Pn3a效能,同时保持对主要脱靶Na V 1.4,Na V 1.5和Na V 1.6大于100倍的选择性。Pn3a [D8N]突变体在体内保留镇痛活性,可明显减轻与临床相关的小鼠术后疼痛的机械异常性疼痛,其剂量比野生型Pn3a低3倍,而不会引起运动不良影响。这项研究的结果将有助于未来合理设计肽类Na V 1.7抑制剂的有效开发,以开发更有效,更安全的止痛药,并进一步研究Na V 1.7在疼痛中的作用。
更新日期:2020-02-19
中文翻译:
绘制止痛药NaV1.7选择性肽Pn3a的分子表面图谱揭示了膜和通道相互作用必不可少的残基。
令人信服的人类遗传研究已经确定了电压门控钠通道Na V 1.7是治疗疼痛的有希望的治疗靶标。镇痛药源自蜘蛛毒液的肽μ-theraphotoxin-Pn3a是Na V 1.7的一种特别有效和选择性的抑制剂。但是,对于定义这种活动的结构-活动关系或渠道相互作用知之甚少。我们合理设计了17种Pn3a类似物,并使用膜片钳电生理学测定了它们在hNa V 1.7时的活性。带有正电荷的氨基酸K22和K24被确定对Pn3a活性至关重要,分子模型确定了这些残基与hNa V结构域II的S3–S4环之间的相互作用1.7。去除疏水残基Y4,Y27和W30会导致效价损失(> 250倍),而用带正电的赖氨酸替代带负电的D1和D8残基会导致效价增加(> 13倍),可能是通过膜脂质相互作用的改变。将D8突变为天冬酰胺可在Na V 1.7(20倍)时最大程度地提高Pn3a效能,同时保持对主要脱靶Na V 1.4,Na V 1.5和Na V 1.6大于100倍的选择性。Pn3a [D8N]突变体在体内保留镇痛活性,可明显减轻与临床相关的小鼠术后疼痛的机械异常性疼痛,其剂量比野生型Pn3a低3倍,而不会引起运动不良影响。这项研究的结果将有助于未来合理设计肽类Na V 1.7抑制剂的有效开发,以开发更有效,更安全的止痛药,并进一步研究Na V 1.7在疼痛中的作用。


























 京公网安备 11010802027423号
京公网安备 11010802027423号