Current Computer-Aided Drug Design ( IF 1.7 ) Pub Date : 2021-03-31 , DOI: 10.2174/1573409916666200219115112 Arijit Bag 1
|
Background: IC50 is one of the most important parameters of a drug. But, it is very difficult to predict this value of a new compound without experiment. There are only a few QSAR based methods available for IC50 prediction, which is also highly dependable on a huge number of known data. Thus, there is an immense demand for a sophisticated computational method of IC50 prediction in the field of in silico drug designing.
Objective: Recently developed quantum computation based method of IC50 prediction by Bag and Ghorai requires an affordable known data. In present research work, further development of this method is carried out such that the requisite number of known data being minimal.
Methods: To retrench the cardinal data span and shrink the effects of variant biological parameters on the computed value of IC50, a relative approach of IC50 computation is pursued in the present method. To predict an approximate value of IC50 of a small molecule, only the IC50 of a similar kind of molecule is required for this method.
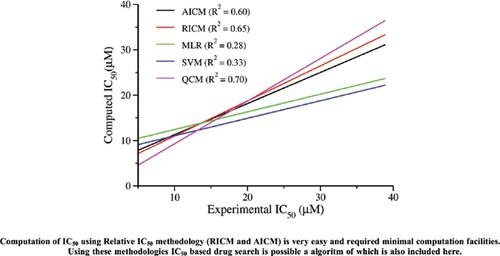
Results: The present method of IC50 computation is tested for both organic and organometallic compounds as HIV-1 capsid A inhibitor and cancer drugs. Computed results match very well with the experiment.
Conclusion: This method is easily applicable to both organic and organometallic compounds with acceptable accuracy. Since this method requires only the dipole moments of an unknown compound and the reference compound, IC50 based drug search is possible with this method. An algorithm is proposed here for IC50 based drug search.
中文翻译:
基于 DFT 的 IC50 预测计算方法
背景:IC 50是药物最重要的参数之一。但是,没有实验就很难预测新化合物的这个值。只有少数基于 QSAR 的方法可用于 IC 50预测,而且这些方法对大量已知数据也非常可靠。因此,在计算机药物设计领域对IC 50预测的复杂计算方法存在巨大需求。
目标:Bag 和 Ghorai最近开发的基于量子计算的 IC 50预测方法需要负担得起的已知数据。在目前的研究工作中,对该方法进行了进一步的开发,使得所需的已知数据数量最少。
方法:为了缩减基本数据跨度并缩小变异生物参数对IC 50计算值的影响,本方法追求IC 50计算的相对方法。为了预测小分子的IC 50的近似值,该方法只需要类似分子的IC 50。
结果: 对作为 HIV-1 衣壳 A 抑制剂和抗癌药物的有机和有机金属化合物测试了本 IC 50计算方法。计算结果与实验非常吻合。
结论:该方法适用于有机和有机金属化合物,准确度可接受。由于此方法只需要未知化合物和参考化合物的偶极矩,因此可以使用此方法进行基于IC 50的药物搜索。这里提出了一种用于基于IC 50的药物搜索的算法。


























 京公网安备 11010802027423号
京公网安备 11010802027423号