Journal of Organometallic Chemistry ( IF 2.3 ) Pub Date : 2020-03-02 , DOI: 10.1016/j.jorganchem.2020.121206 Daniel Zárate-Saldaña , Bruno Landeros-Rivera , Jorge A. Cruz-Morales , Selena Gutiérrez
|
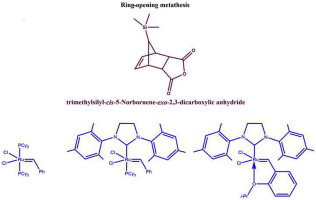
The monomer synthesis and ring-opening metathesis polymerization (ROMP) of cis-5-norbornene-exo-2,3-dicarboxylic anhydride (1a) and 7-syn-trimethylsilyl-cis-5-norbornene-exo-2,3-dicarboxylic anhydride (1b) mediated by ruthenium-alkylidene catalysts (I, II and III) were experimentally carried out. Metathesis reaction pathways of 1a and 1b monomers using II have been studied at PBE-D3(BJ)/def2-TZVP level of theory, employing the SMD model for simulation of 1,2-dichloroethane solvent. The calculations unravel that reactivity difference between 1a and 1b towards ruthenium alkylidene complex II is due to the fact that the intermediate π-complex formation was found in the 1a reaction pathway but was absent in the 1b one. Moreover, there are marked differences in the formation processes of the metallacyclobutane intermediaries 5a and 5b, the first is an exergonic process (−8.3 kcal/mol) and the last one is an endergonic process (2 kcal/mol), in addition to the high activation energy of the monomer 1b (15.8 kcal/mol) compared with 1a (5.5 kcal/mol). Such differences are attributed to the high steric impediment imposed by -Si(CH3)3 over the double bond (syn conformation). Using quantum theory of atoms in molecules (QTAIM) it was possible to analyze successfully the mechanistic pathway of metathesis reaction for both monomers, complementing the results obtained by DFT energetic analysis.
中文翻译:
Ru-亚烷基催化剂对含三甲基甲硅烷基的降冰片烯衍生物的复分解:实验和计算研究
顺式-5-降冰片烯-exo -2,3-二羧酸酐(1a)和7-顺-三甲基甲硅烷基-顺式-5-降冰片烯-exo -2,3-二羧酸的单体合成和开环复分解聚合(ROMP)实验性地进行了由钌-亚烷基催化剂(I,II和III)介导的酸酐(1b)。II使用1a和1b单体进行复分解反应的途径已经在PBE-D3(BJ)/ def2-TZVP的理论水平上进行了研究,采用SMD模型来模拟1,2-二氯乙烷溶剂。该计算揭示了1a和1b之间对钌亚烷基络合物II的反应性差异是由于以下事实:在1a反应途径中发现了中间的π-络合物形成,但在1b反应途径中却没有。而且,金属环环丁烷中间体5a和5b的形成过程存在明显差异。,首先是一个放能过程(-8.3千卡/摩尔)和最后一个是吸能过程(2千卡/摩尔),除了单体的高活化能1B(15.8千卡/ mol)与比较1A( 5.5 kcal / mol)。此类差异归因于-Si(CH 3)3对双键(顺式构象)施加的高位阻。使用分子中原子的量子理论(QTAIM),可以成功地分析两种单体的复分解反应的机理,并补充通过DFT能量分析获得的结果。


























 京公网安备 11010802027423号
京公网安备 11010802027423号