当前位置:
X-MOL 学术
›
J. Mol. Biol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Sequence-Based Prediction of Fuzzy Protein Interactions.
Journal of Molecular Biology ( IF 5.6 ) Pub Date : 2020-02-27 , DOI: 10.1016/j.jmb.2020.02.017 Marton Miskei 1 , Attila Horvath 2 , Michele Vendruscolo 3 , Monika Fuxreiter 1
Journal of Molecular Biology ( IF 5.6 ) Pub Date : 2020-02-27 , DOI: 10.1016/j.jmb.2020.02.017 Marton Miskei 1 , Attila Horvath 2 , Michele Vendruscolo 3 , Monika Fuxreiter 1
Affiliation
|
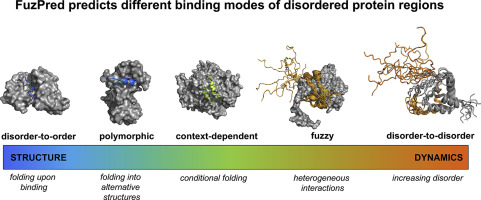
It is becoming increasingly recognised that disordered proteins may be fuzzy, in that they can exhibit a wide variety of binding modes. In addition to the well-known process of folding upon binding (disorder-to-order transition), many examples are emerging of interacting proteins that remain disordered in their bound states (disorder-to-disorder transitions). Furthermore, disordered proteins may populate ordered and disordered states to different extents depending on their partners (context-dependent binding). Here we assemble three datasets comprising disorder-to-order, context-dependent, and disorder-to-disorder transitions of 828 protein regions represented in 2157 complexes and elucidate the sequence-determinants of the different interaction modes. We found that fuzzy interactions originate from local sequence compositions that promote the sampling of a wide range of different structures. Based on this observation, we developed the FuzPred method (http://protdyn-fuzpred.org) of predicting the binding modes of disordered proteins based on their amino acid sequences, without specifying their partners. We thus illustrate how the amino acid sequences of proteins can encode a wide range of conformational changes upon binding, including transitions from disordered to ordered and from disordered to disordered states.
中文翻译:

模糊蛋白质相互作用的基于序列的预测。
越来越多地认识到无序蛋白可能是模糊的,因为它们可以表现出多种结合模式。除了众所周知的结合时折叠的过程(无序过渡),许多相互作用蛋白的例子也不断出现,这些蛋白在其结合状态(无序过渡)保持无序。此外,无序蛋白可能会根据其配偶体(上下文依赖性结合)而在不同程度上填充有序状态和无序状态。在这里,我们组装了三个数据集,包括2157个复合体中表示的828个蛋白区域的无序排列,上下文相关和无序过渡,并阐明了不同相互作用模式的序列决定因素。我们发现模糊的相互作用源于局部序列组成,其促进了各种不同结构的采样。基于此观察,我们开发了FuzPred方法(http://protdyn-fuzpred.org),该方法可根据无序蛋白的氨基酸序列预测无序蛋白的结合模式,而无需指定其伴侣。因此,我们说明了蛋白质的氨基酸序列如何在结合后编码广泛的构象变化,包括从无序到有序以及从无序到无序状态的转变。
更新日期:2020-02-27
中文翻译:
模糊蛋白质相互作用的基于序列的预测。
越来越多地认识到无序蛋白可能是模糊的,因为它们可以表现出多种结合模式。除了众所周知的结合时折叠的过程(无序过渡),许多相互作用蛋白的例子也不断出现,这些蛋白在其结合状态(无序过渡)保持无序。此外,无序蛋白可能会根据其配偶体(上下文依赖性结合)而在不同程度上填充有序状态和无序状态。在这里,我们组装了三个数据集,包括2157个复合体中表示的828个蛋白区域的无序排列,上下文相关和无序过渡,并阐明了不同相互作用模式的序列决定因素。我们发现模糊的相互作用源于局部序列组成,其促进了各种不同结构的采样。基于此观察,我们开发了FuzPred方法(http://protdyn-fuzpred.org),该方法可根据无序蛋白的氨基酸序列预测无序蛋白的结合模式,而无需指定其伴侣。因此,我们说明了蛋白质的氨基酸序列如何在结合后编码广泛的构象变化,包括从无序到有序以及从无序到无序状态的转变。

























 京公网安备 11010802027423号
京公网安备 11010802027423号