当前位置:
X-MOL 学术
›
Nat. Protoc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Measurement of differential chromatin interactions with absolute quantification of architecture (AQuA-HiChIP).
Nature Protocols ( IF 14.8 ) Pub Date : 2020-02-12 , DOI: 10.1038/s41596-019-0285-9 Berkley E Gryder 1 , Javed Khan 1 , Benjamin Z Stanton 2
Nature Protocols ( IF 14.8 ) Pub Date : 2020-02-12 , DOI: 10.1038/s41596-019-0285-9 Berkley E Gryder 1 , Javed Khan 1 , Benjamin Z Stanton 2
Affiliation
|
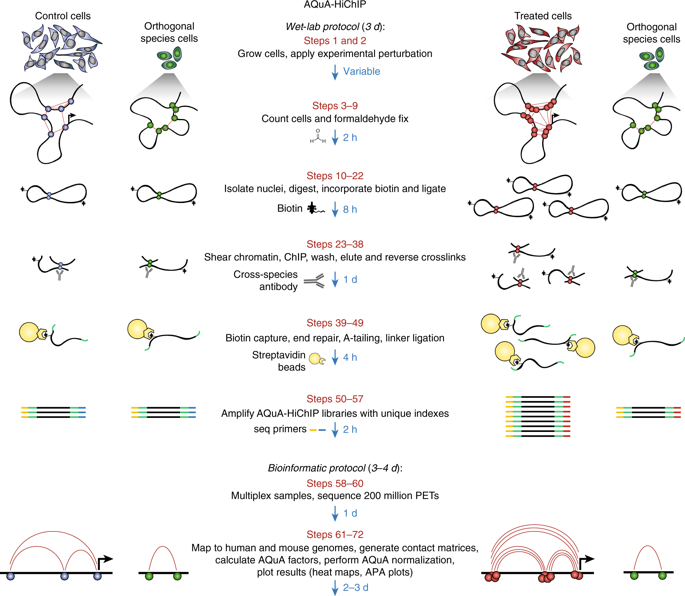
Methods developed to capture protein-anchored chromatin interactions (chromatin interaction analysis by paired-end tag sequencing and HiChIP) have yielded tremendous insights into the 3D folding principles of the genome, but are normalized by sequencing depth and therefore unable to accurately measure global changes in chromatin interactions and contact domain organization. We herein describe the protocol for absolute quantification of chromatin architecture (AQuA)-HiChIP, an advance that allows the absolute differences in protein-anchored chromatin interactions between samples to be determined. With our method, defined ratios of mouse and human fixed nuclei are mixed and subjected to endonuclease digestion. Chromatin contacts are captured by biotin-dATP incorporation and proximity ligation, followed by gentle shearing, ChIP, biotin capture and paired-end sequencing. 3D contacts are counted from paired-end tags (PETs) from the human genome and are normalized to the total PETs from the mouse genome. As orthogonal normalization allows observation of global changes, the approach will enable more quantitative insights into the topological determinants of transcriptional control and tissue-specific epigenetic memory. With our approach, we have discovered that rapid histone deacetylase inhibition disrupts super enhancer function by creating many new aberrant contacts. The code for data analysis is available at https://github.com/GryderArt/AQuA-HiChIP. This protocol reports both experimental and bioinformatic details to perform AQuA-HiChIP, going from cell culture to ranking chromatin interactions within 6 d.
中文翻译:

使用绝对定量架构(AQuA-HiChIP)测量差异染色质相互作用。
为捕获蛋白质锚定的染色质相互作用而开发的方法(通过配对末端标签测序和HiChIP进行染色质相互作用分析)已对基因组的3D折叠原理产生了深刻的见解,但已通过测序深度进行了归一化,因此无法准确测量染色质相互作用和接触域的组织。我们在本文中描述了用于染色质结构(AQuA)-HiChIP的绝对定量的协议,该进展允许确定样品之间蛋白质锚定的染色质相互作用的绝对差异。用我们的方法,将确定比例的小鼠和人固定核混合并进行核酸内切酶消化。染色质接触是通过生物素-dATP掺入和邻近结扎,然后轻轻剪切,ChIP,生物素捕获和配对末端测序。从人类基因组的配对末端标签(PET)对3D接触进行计数,并根据小鼠基因组的总PET进行标准化。由于正交归一化可以观察到全局变化,因此该方法将对转录控制和组织特异性表观遗传记忆的拓扑决定因素提供更多的定量见解。通过我们的方法,我们发现快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。从人类基因组的配对末端标签(PET)对3D接触进行计数,并根据小鼠基因组的总PET进行标准化。由于正交归一化可以观察到全局变化,因此该方法将对转录控制和组织特异性表观遗传记忆的拓扑决定因素提供更多的定量见解。通过我们的方法,我们发现快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。从人类基因组的配对末端标签(PET)对3D接触进行计数,并根据小鼠基因组的总PET进行标准化。由于正交归一化可以观察到全局变化,因此该方法将对转录控制和组织特异性表观遗传记忆的拓扑决定因素提供更多的定量见解。通过我们的方法,我们发现快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。由于正交归一化可以观察到全局变化,因此该方法将对转录控制和组织特异性表观遗传记忆的拓扑决定因素提供更多的定量见解。通过我们的方法,我们发现快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。由于正交归一化可以观察到全局变化,因此该方法将对转录控制和组织特异性表观遗传记忆的拓扑决定因素提供更多的定量见解。通过我们的方法,我们发现快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。我们发现,快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子的功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。我们发现,快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子的功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。
更新日期:2020-02-12
中文翻译:
使用绝对定量架构(AQuA-HiChIP)测量差异染色质相互作用。
为捕获蛋白质锚定的染色质相互作用而开发的方法(通过配对末端标签测序和HiChIP进行染色质相互作用分析)已对基因组的3D折叠原理产生了深刻的见解,但已通过测序深度进行了归一化,因此无法准确测量染色质相互作用和接触域的组织。我们在本文中描述了用于染色质结构(AQuA)-HiChIP的绝对定量的协议,该进展允许确定样品之间蛋白质锚定的染色质相互作用的绝对差异。用我们的方法,将确定比例的小鼠和人固定核混合并进行核酸内切酶消化。染色质接触是通过生物素-dATP掺入和邻近结扎,然后轻轻剪切,ChIP,生物素捕获和配对末端测序。从人类基因组的配对末端标签(PET)对3D接触进行计数,并根据小鼠基因组的总PET进行标准化。由于正交归一化可以观察到全局变化,因此该方法将对转录控制和组织特异性表观遗传记忆的拓扑决定因素提供更多的定量见解。通过我们的方法,我们发现快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。从人类基因组的配对末端标签(PET)对3D接触进行计数,并根据小鼠基因组的总PET进行标准化。由于正交归一化可以观察到全局变化,因此该方法将对转录控制和组织特异性表观遗传记忆的拓扑决定因素提供更多的定量见解。通过我们的方法,我们发现快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。从人类基因组的配对末端标签(PET)对3D接触进行计数,并根据小鼠基因组的总PET进行标准化。由于正交归一化可以观察到全局变化,因此该方法将对转录控制和组织特异性表观遗传记忆的拓扑决定因素提供更多的定量见解。通过我们的方法,我们发现快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。由于正交归一化可以观察到全局变化,因此该方法将对转录控制和组织特异性表观遗传记忆的拓扑决定因素提供更多的定量见解。通过我们的方法,我们发现快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。由于正交归一化可以观察到全局变化,因此该方法将对转录控制和组织特异性表观遗传记忆的拓扑决定因素提供更多的定量见解。通过我们的方法,我们发现快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。我们发现,快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子的功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。我们发现,快速的组蛋白脱乙酰基酶抑制作用通过产生许多新的异常接触而破坏了超级增强子的功能。数据分析代码可从https://github.com/GryderArt/AQuA-HiChIP获得。该协议报告了执行AQuA-HiChIP的实验和生物信息学细节,从细胞培养到6 d内染色质相互作用排名。

























 京公网安备 11010802027423号
京公网安备 11010802027423号