当前位置:
X-MOL 学术
›
Arch. Biochem. Biophys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Endoplasmic reticulum stress-induced complex I defect: Central role of calcium overload.
Archives of Biochemistry and Biophysics ( IF 3.9 ) Pub Date : 2020-02-12 , DOI: 10.1016/j.abb.2020.108299 Ahmed A Mohsin 1 , Jeremy Thompson 2 , Ying Hu 2 , John Hollander 3 , Edward J Lesnefsky 4 , Qun Chen 2
Archives of Biochemistry and Biophysics ( IF 3.9 ) Pub Date : 2020-02-12 , DOI: 10.1016/j.abb.2020.108299 Ahmed A Mohsin 1 , Jeremy Thompson 2 , Ying Hu 2 , John Hollander 3 , Edward J Lesnefsky 4 , Qun Chen 2
Affiliation
|
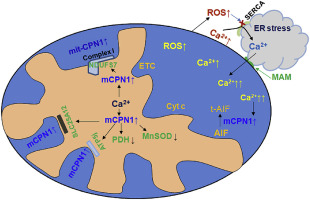
BACKGROUND
ER (endoplasmic reticulum) stress leads to decreased complex I activity in cardiac mitochondria. The aim of the current study is to explore the potential mechanisms by which ER stress leads to the complex I defect. ER stress contributes to intracellular calcium overload and oxidative stress that are two key factors to induce mitochondrial dysfunction. Since oxidative stress is often accompanied by intracellular calcium overload during ER stress in vivo, the role of oxidative stress and calcium overload in mitochondrial dysfunction was studied using in vitro models. ER stress results in intracellular calcium overload that favors activation of calcium-dependent calpains. The contribution of mitochondrial calpain activation in ER stress-mediated complex I damage was studied.
METHODS
Thapsigargin (THAP) was used to induce acute ER stress in H9c2 cells and C57BL/6 mice. Exogenous calcium (25 μM) and H2O2 (100 μM) were used to induce modest calcium overload and oxidative stress in isolated mitochondria. Calpain small subunit 1 (CAPNS1) is essential to maintain calpain 1 and calpain 2 (CPN1/2) activities. Deletion of CAPNS1 eliminates the activities of CPN1/2. Wild type and cardiac-specific CAPNS1 deletion mice were used to explore the role of CPN1/2 activation in calcium-induced mitochondrial damage.
RESULTS
In isolated mitochondria, exogenous calcium but not H2O2 treatment led to decreased oxidative phosphorylation, supporting that calcium overload contributes a key role in the mitochondrial damage. THAP treatment of H9c2 cells decreased respiration selectively with complex I substrates. THAP treatment activated cytosolic and mitochondrial CPN1/2 in C57BL/6 mice and led to degradation of complex I subunits including NDUFS7. Calcium treatment decreased NDUFS7 content in wild type but not in CAPNS1 knockout mice.
CONCLUSION
ER stress-mediated activation of mitochondria-localized CPN1/2 contributes to complex I damage by cleaving component subunits.
中文翻译:

内质网应激诱导的复合物 I 缺陷:钙超载的中心作用。
背景 ER(内质网)应激导致心脏线粒体中复合物 I 活性降低。目前研究的目的是探索 ER 应力导致复杂 I 缺陷的潜在机制。ER 应激导致细胞内钙超载和氧化应激,这是诱导线粒体功能障碍的两个关键因素。由于体内 ER 应激期间氧化应激通常伴随细胞内钙超载,因此使用体外模型研究了氧化应激和钙超载在线粒体功能障碍中的作用。ER 应激导致细胞内钙超载,有利于激活钙依赖性钙蛋白酶。研究了线粒体钙蛋白酶激活在 ER 应激介导的复合物 I 损伤中的作用。方法毒胡萝卜素 (THAP) 用于在 H9c2 细胞和 C57BL/6 小鼠中诱导急性 ER 应激。外源性钙 (25 μM) 和 H2O2 (100 μM) 用于在分离的线粒体中诱导适度的钙超载和氧化应激。钙蛋白酶小亚基 1 (CAPNS1) 是维持钙蛋白酶 1 和钙蛋白酶 2 (CPN1/2) 活动所必需的。CAPNS1 的删除消除了 CPN1/2 的活动。野生型和心脏特异性 CAPNS1 缺失小鼠用于探索 CPN1/2 激活在钙诱导的线粒体损伤中的作用。结果 在分离的线粒体中,外源性钙而非 H2O2 处理导致氧化磷酸化降低,支持钙超载在线粒体损伤中起关键作用。H9c2 细胞的 THAP 处理选择性地降低了复杂 I 底物的呼吸。THAP 处理激活 C57BL/6 小鼠的细胞溶质和线粒体 CPN1/2,并导致包括 NDUFS7 在内的复杂 I 亚基降解。钙处理降低了野生型中 NDUFS7 的含量,但在 CAPNS1 敲除小鼠中没有。结论 ER 应激介导的线粒体定位 CPN1/2 激活通过切割组分亚基导致复合物 I 损伤。
更新日期:2020-02-12
中文翻译:
内质网应激诱导的复合物 I 缺陷:钙超载的中心作用。
背景 ER(内质网)应激导致心脏线粒体中复合物 I 活性降低。目前研究的目的是探索 ER 应力导致复杂 I 缺陷的潜在机制。ER 应激导致细胞内钙超载和氧化应激,这是诱导线粒体功能障碍的两个关键因素。由于体内 ER 应激期间氧化应激通常伴随细胞内钙超载,因此使用体外模型研究了氧化应激和钙超载在线粒体功能障碍中的作用。ER 应激导致细胞内钙超载,有利于激活钙依赖性钙蛋白酶。研究了线粒体钙蛋白酶激活在 ER 应激介导的复合物 I 损伤中的作用。方法毒胡萝卜素 (THAP) 用于在 H9c2 细胞和 C57BL/6 小鼠中诱导急性 ER 应激。外源性钙 (25 μM) 和 H2O2 (100 μM) 用于在分离的线粒体中诱导适度的钙超载和氧化应激。钙蛋白酶小亚基 1 (CAPNS1) 是维持钙蛋白酶 1 和钙蛋白酶 2 (CPN1/2) 活动所必需的。CAPNS1 的删除消除了 CPN1/2 的活动。野生型和心脏特异性 CAPNS1 缺失小鼠用于探索 CPN1/2 激活在钙诱导的线粒体损伤中的作用。结果 在分离的线粒体中,外源性钙而非 H2O2 处理导致氧化磷酸化降低,支持钙超载在线粒体损伤中起关键作用。H9c2 细胞的 THAP 处理选择性地降低了复杂 I 底物的呼吸。THAP 处理激活 C57BL/6 小鼠的细胞溶质和线粒体 CPN1/2,并导致包括 NDUFS7 在内的复杂 I 亚基降解。钙处理降低了野生型中 NDUFS7 的含量,但在 CAPNS1 敲除小鼠中没有。结论 ER 应激介导的线粒体定位 CPN1/2 激活通过切割组分亚基导致复合物 I 损伤。

























 京公网安备 11010802027423号
京公网安备 11010802027423号