当前位置:
X-MOL 学术
›
Arch. Pharm.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Design, synthesis, and molecular docking of novel 2‐arylbenzothiazole multiangiokinase inhibitors targeting breast cancer
Archiv der Pharmazie ( IF 5.1 ) Pub Date : 2020-04-01 , DOI: 10.1002/ardp.201900340 Heba T Abdel-Mohsen 1 , Eman A Abd El-Meguid 1 , Ahmed M El Kerdawy 2, 3 , Abeer E E Mahmoud 4 , Mamdouh M Ali 4
Archiv der Pharmazie ( IF 5.1 ) Pub Date : 2020-04-01 , DOI: 10.1002/ardp.201900340 Heba T Abdel-Mohsen 1 , Eman A Abd El-Meguid 1 , Ahmed M El Kerdawy 2, 3 , Abeer E E Mahmoud 4 , Mamdouh M Ali 4
Affiliation
|
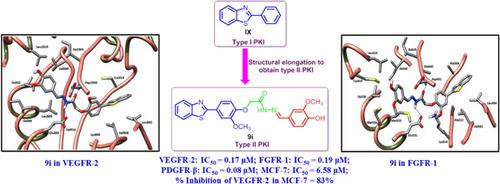
A novel series of 2‐arylbenzothiazoles 9, 10, and 12 were designed and synthesized as VEGFR‐2/FGFR‐1/PDGFR‐β multiangiokinase inhibitors targeting breast cancer. Structural elongation of the known 2‐phenylbenzothiazole scaffold (type I protein kinase inhibitor [PKI]), was carried out to afford series of type II PKIs 9, 10, and 12. Compounds 9d, 9f, 9i, and 9k exhibited potent multikinase inhibitory activity with IC50 values of 0.19, 0.18, 0.17, and 0.13 μM, respectively, against VEGFR‐2; IC50 values of 0.28, 0.37, 0.19, and 0.27 μM, respectively, against FGFR‐1; and IC50 values of 0.07, 0.04, 0.08, and 0.14 μM, respectively, against PDGFR‐β. Moreover, the synthesized benzothiazoles demonstrated promising cytotoxic activity against the MCF‐7 cell line. The most potent benzothiazoles 9d and 9i exhibited IC50 values of 7.83 and 6.58 μM, respectively, on the MCF‐7 cell line in comparison to sorafenib (III), which showed IC50 = 4.33 μM. Additionally, 9d and 9i showed VEGFR‐2 inhibitory activity in MCF‐7 cells of 81% and 83% when compared with sorafenib (III), which showed 88% inhibition. Molecular docking of the designed compounds in the VEGFR‐2 and FGFR‐1 active sites showed the accommodation of the 2‐phenylbenzothiazole moiety, as reported, in the hinge region of the receptor tyrosine kinase (RTK)‐binding site, while the amide moiety is involved in hydrogen bond interactions with the key amino acids in the gate area; this in turn directs the aryl group to the hydrophobic allosteric back pocket of the RTKs in a type II‐like binding mode. The synthesized benzothiazoles showed satisfactory ADME properties for further optimization in drug discovery.
中文翻译:

靶向乳腺癌的新型2-芳基苯并噻唑多血管激酶抑制剂的设计、合成和分子对接
设计并合成了一系列新的 2-芳基苯并噻唑 9、10 和 12,作为靶向乳腺癌的 VEGFR-2/FGFR-1/PDGFR-β 多血管激酶抑制剂。对已知的 2-苯基苯并噻唑支架(I 型蛋白激酶抑制剂 [PKI])进行结构延伸,得到一系列 II 型 PKI 9、10 和 12。化合物 9d、9f、9i 和 9k 表现出强效的多激酶抑制作用对 VEGFR-2 的 IC50 值分别为 0.19、0.18、0.17 和 0.13 μM;对 FGFR-1 的 IC50 值分别为 0.28、0.37、0.19 和 0.27 μM;对 PDGFR-β 的 IC50 和 IC50 值分别为 0.07、0.04、0.08 和 0.14 μM。此外,合成的苯并噻唑对 MCF-7 细胞系显示出有希望的细胞毒活性。最有效的苯并噻唑 9d 和 9i 的 IC50 值分别为 7.83 和 6.58 μM,在 MCF-7 细胞系上与索拉非尼 (III) 相比,IC50 = 4.33 μM。此外,与显示 88% 抑制的索拉非尼 (III) 相比,9d 和 9i 在 MCF-7 细胞中显示出 81% 和 83% 的 VEGFR-2 抑制活性。设计的化合物在 VEGFR-2 和 FGFR-1 活性位点的分子对接显示,在受体酪氨酸激酶 (RTK) 结合位点的铰链区调节了 2-苯基苯并噻唑部分,而酰胺部分参与与门区关键氨基酸的氢键相互作用;这反过来以类 II 型结合模式将芳基引导至 RTK 的疏水变构后袋。合成的苯并噻唑显示出令人满意的 ADME 特性,可用于药物发现的进一步优化。
更新日期:2020-04-01
中文翻译:
靶向乳腺癌的新型2-芳基苯并噻唑多血管激酶抑制剂的设计、合成和分子对接
设计并合成了一系列新的 2-芳基苯并噻唑 9、10 和 12,作为靶向乳腺癌的 VEGFR-2/FGFR-1/PDGFR-β 多血管激酶抑制剂。对已知的 2-苯基苯并噻唑支架(I 型蛋白激酶抑制剂 [PKI])进行结构延伸,得到一系列 II 型 PKI 9、10 和 12。化合物 9d、9f、9i 和 9k 表现出强效的多激酶抑制作用对 VEGFR-2 的 IC50 值分别为 0.19、0.18、0.17 和 0.13 μM;对 FGFR-1 的 IC50 值分别为 0.28、0.37、0.19 和 0.27 μM;对 PDGFR-β 的 IC50 和 IC50 值分别为 0.07、0.04、0.08 和 0.14 μM。此外,合成的苯并噻唑对 MCF-7 细胞系显示出有希望的细胞毒活性。最有效的苯并噻唑 9d 和 9i 的 IC50 值分别为 7.83 和 6.58 μM,在 MCF-7 细胞系上与索拉非尼 (III) 相比,IC50 = 4.33 μM。此外,与显示 88% 抑制的索拉非尼 (III) 相比,9d 和 9i 在 MCF-7 细胞中显示出 81% 和 83% 的 VEGFR-2 抑制活性。设计的化合物在 VEGFR-2 和 FGFR-1 活性位点的分子对接显示,在受体酪氨酸激酶 (RTK) 结合位点的铰链区调节了 2-苯基苯并噻唑部分,而酰胺部分参与与门区关键氨基酸的氢键相互作用;这反过来以类 II 型结合模式将芳基引导至 RTK 的疏水变构后袋。合成的苯并噻唑显示出令人满意的 ADME 特性,可用于药物发现的进一步优化。

























 京公网安备 11010802027423号
京公网安备 11010802027423号