当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Lipopolysaccharide Simulations Are Sensitive to Phosphate Charge and Ion Parameterization.
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-02-26 , DOI: 10.1021/acs.jctc.9b00868 Amy Rice 1 , Mary T Rooney 2 , Alexander I Greenwood 2, 3 , Myriam L Cotten 2 , Jeff Wereszczynski 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-02-26 , DOI: 10.1021/acs.jctc.9b00868 Amy Rice 1 , Mary T Rooney 2 , Alexander I Greenwood 2, 3 , Myriam L Cotten 2 , Jeff Wereszczynski 1
Affiliation
|
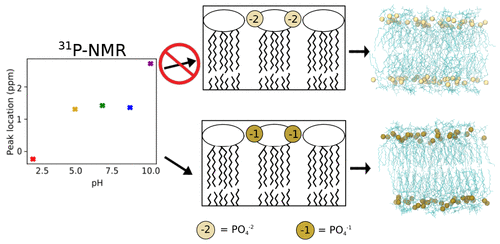
The high proportion of lipopolysaccharide (LPS) molecules in the outer membrane of Gram-negative bacteria makes it a highly effective barrier to small molecules, antibiotic drugs, and other antimicrobial agents. Given this vital role in protecting bacteria from potentially hostile environments, simulations of LPS bilayers and outer membrane systems represent a critical tool for understanding the mechanisms of bacterial resistance and the development of new antibiotic compounds that circumvent these defenses. The basis of these simulations is parameterizations of LPS, which have been developed for all major molecular dynamics force fields. However, these parameterizations differ in both the protonation state of LPS and how LPS membranes behave in the presence of various ion species. To address these discrepancies and understand the effects of phosphate charge on bilayer properties, simulations were performed for multiple distinct LPS chemotypes with different ion parameterizations in both protonated or deprotonated lipid A states. These simulations show that bilayer properties, such as the area per lipid and inter-lipid hydrogen bonding, are highly influenced by the choice of phosphate group charges, cation type, and ion parameterization, with protonated LPS and monovalent cations with modified nonbonded parameters providing the best match to the experiments. Additionally, alchemical free energy simulations were performed to determine theoretical pKa values for LPS and subsequently validated by 31P solid-state nuclear magnetic resonance experiments. Results from these complementary computational and experimental studies demonstrate that the protonated state dominates at physiological pH, contrary to the deprotonated form modeled by many LPS force fields. Overall, these results highlight the sensitivity of LPS simulations to phosphate charge and ion parameters while offering recommendations for how existing models should be updated for consistency between force fields as well as to best match experiments.
中文翻译:

脂多糖模拟对磷酸盐电荷和离子参数化敏感。
革兰氏阴性细菌外膜中脂多糖(LPS)分子的比例很高,使其成为小分子,抗生素药物和其他抗菌剂的高效屏障。鉴于在保护细菌免受潜在敌对环境的影响中起着至关重要的作用,LPS双层和外膜系统的仿真代表了理解细菌耐药性机制和开发可克服这些防御作用的新型抗生素化合物的关键工具。这些模拟的基础是LPS的参数化,已针对所有主要的分子动力学力场进行了开发。但是,这些参数化在LPS的质子化状态和LPS膜在各种离子种类存在下的行为方面都不同。为了解决这些差异并了解磷酸盐电荷对双层性能的影响,针对质子化或去质子化脂质A状态下具有不同离子参数化的多种不同LPS化学型进行了仿真。这些模拟表明,双层性质(例如每个脂质的面积和脂质间的氢键结合)受磷酸根基团电荷选择,阳离子类型和离子参数化的影响很大,质子化的LPS和单价阳离子的修饰非键合参数可提供最适合实验。此外,进行了炼金术自由能模拟,以确定LPS的理论pKa值,随后通过31P固态核磁共振实验进行了验证。这些互补的计算和实验研究的结果表明,质子化状态在生理pH下占主导地位,这与许多LPS力场所模拟的去质子化形式相反。总体而言,这些结果突出了LPS模拟对磷酸盐电荷和离子参数的敏感性,同时为如何更新现有模型以提供力场之间的一致性以及最佳匹配实验提供了建议。
更新日期:2020-02-27
中文翻译:
脂多糖模拟对磷酸盐电荷和离子参数化敏感。
革兰氏阴性细菌外膜中脂多糖(LPS)分子的比例很高,使其成为小分子,抗生素药物和其他抗菌剂的高效屏障。鉴于在保护细菌免受潜在敌对环境的影响中起着至关重要的作用,LPS双层和外膜系统的仿真代表了理解细菌耐药性机制和开发可克服这些防御作用的新型抗生素化合物的关键工具。这些模拟的基础是LPS的参数化,已针对所有主要的分子动力学力场进行了开发。但是,这些参数化在LPS的质子化状态和LPS膜在各种离子种类存在下的行为方面都不同。为了解决这些差异并了解磷酸盐电荷对双层性能的影响,针对质子化或去质子化脂质A状态下具有不同离子参数化的多种不同LPS化学型进行了仿真。这些模拟表明,双层性质(例如每个脂质的面积和脂质间的氢键结合)受磷酸根基团电荷选择,阳离子类型和离子参数化的影响很大,质子化的LPS和单价阳离子的修饰非键合参数可提供最适合实验。此外,进行了炼金术自由能模拟,以确定LPS的理论pKa值,随后通过31P固态核磁共振实验进行了验证。这些互补的计算和实验研究的结果表明,质子化状态在生理pH下占主导地位,这与许多LPS力场所模拟的去质子化形式相反。总体而言,这些结果突出了LPS模拟对磷酸盐电荷和离子参数的敏感性,同时为如何更新现有模型以提供力场之间的一致性以及最佳匹配实验提供了建议。

























 京公网安备 11010802027423号
京公网安备 11010802027423号