Journal of Molecular Liquids ( IF 6 ) Pub Date : 2020-01-27 , DOI: 10.1016/j.molliq.2020.112581 Gokhan Kacar , Gijsbertus de With
|
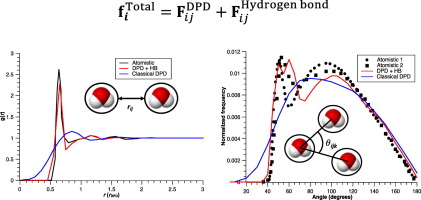
Simulating water has always been a challenge. Due to the intrinsic hydrogen bond interactions, water exhibits structural properties, such as a tetrahedral coordination resulting in a specific Radial Distribution Function (RDF), which are not trivial to predict computationally. In this paper, we attempt to use coarse-grained Dissipative Particle Dynamics (DPD) simulations to parameterize the hydrogen bond interactions without violating the classical DPD framework. We model the hydrogen bond interactions by incorporating a Morse potential, where the parameters are computed by taking the experimental enthalpy of evaporation and hydrogen bond distances as reference. We show that with the proposed procedure the RDF, the coordination number, the isothermal compressibility, and the three-body angular distributions (to demonstrate the tetrahedral structure) of pure water are predicted in great extent compatible with the experiments. To test the applicability of the procedure to mixtures, we simulated pure methanol and methanol/water mixtures at different molar fractions. The predicted RDF profiles for methanol-methanol, methanol-water and water-water represent the characteristic experimental RDF behavior. Moreover, the calculated negative excess volumes as a function of mole fraction compare quite well with the experimentally observed excess volumes. Our findings motivate the further development and use of DPD simulations in modeling hydrogen bond interactions, which are crucial not only in water (or alcohols), but in more complex systems such as biomolecules, proteins or biopolymers.
中文翻译:
在耗散粒子动力学模拟中参数化氢键相互作用:以水,甲醇及其二元混合物为例
模拟水一直是一个挑战。由于固有的氢键相互作用,水表现出结构特性,例如四面体配位,从而产生特定的径向分布函数(RDF),这在计算上并不容易。在本文中,我们尝试使用粗粒度的耗散粒子动力学(DPD)模拟来设定氢键相互作用的参数,而不违反经典的DPD框架。我们通过结合莫尔斯电势对氢键相互作用进行建模,其中参数是通过将蒸发的实验焓和氢键距离作为参考来计算的。我们证明,通过提出的程序,RDF,配位数,等温压缩率,预测纯水的三体角分布(以证明四面体结构)在很大程度上与实验兼容。为了测试该程序对混合物的适用性,我们模拟了不同摩尔分数的纯甲醇和甲醇/水混合物。预测的甲醇-甲醇,甲醇-水和水-水的RDF曲线代表了典型的实验RDF行为。而且,作为摩尔分数的函数计算的负过量体积与实验观察到的过量体积相当好。我们的发现激励了DPD模拟在氢键相互作用建模中的进一步发展和使用,这不仅在水(或醇)中,而且在更复杂的系统(例如生物分子,蛋白质或生物聚合物)中也至关重要。


























 京公网安备 11010802027423号
京公网安备 11010802027423号