当前位置:
X-MOL 学术
›
J. Heterocycl. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Mechanistic study of the tandem intramolecular (4 + 2)/intermolecular (3 + 2) cycloaddition reactions for the formation of polyaza‐ and polyisoxazolidine‐steroids
Journal of Heterocyclic Chemistry ( IF 2.4 ) Pub Date : 2020-01-24 , DOI: 10.1002/jhet.3900 Ernest Opoku 1 , George Baffour Pipim 1 , Richard Tia 1 , Evans Adei 1
Journal of Heterocyclic Chemistry ( IF 2.4 ) Pub Date : 2020-01-24 , DOI: 10.1002/jhet.3900 Ernest Opoku 1 , George Baffour Pipim 1 , Richard Tia 1 , Evans Adei 1
Affiliation
|
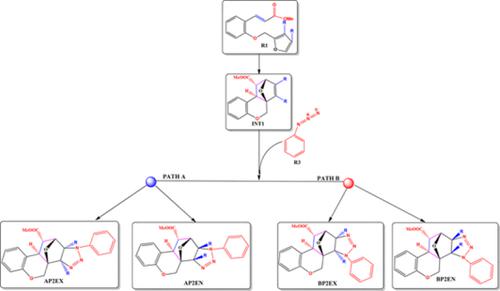
The ability to construct molecules with potential applications in biomedicine via efficient and selective molecular design and syntheses hinges on a thorough understanding of underlying reaction mechanisms. The biological importance of steroids and related heterocyclic compounds are well known but theoretical studies aimed at delineating reaction mechanisms to complement efforts of experimentalists are lacking. Herein, we report an extensive theoretical study on the regio‐, stereo‐, and enantio‐selectivity of the tandem sequential intramolecular (4 + 2)/intermolecular (3 + 2) cycloaddition reaction of (E)‐3‐(2‐[furan‐2‐ylmethoxy]phenyl)acrylate derivatives (R1) and azides (R3 and R3′) as well as nitrones (R2) for the formation of steroids. In the reaction of R1 and the azide derivatives R3 and R3′, the intramolecular (4 + 2) cycloaddition of R1 is the rate‐determining step (rds). The reaction is also found to be very selective. Reaction of electron‐withdrawing groups‐substituted R1 with R3 is found to generally increase the barrier of the rds except bromine whiles electron‐donating groups are found to generally decrease the activation energies of the rds. Subsequently, we report a novel reaction of R1 with cyclic nitrone (R2), which compares favorably with the azide reaction. Results from the global reactivity descriptors are in good agreement with the activation barrier trends. All the considered reactions in this study are found to be kinetically driven.
中文翻译:

串联分子内(4 + 2)/分子间(3 + 2)环加成反应形成聚氮杂和聚异恶唑烷类固醇的机理研究
通过有效和选择性的分子设计和合成来构建在生物医学中具有潜在应用潜力的分子的能力取决于对潜在反应机制的透彻了解。类固醇和相关杂环化合物的生物学重要性是众所周知的,但是缺乏旨在描述反应机理以补充实验者的努力的理论研究。本文中,我们报道了有关(E)-3-(2-[[]]的串联顺序分子内(4 + 2)/分子间(3 + 2)环加成反应的区域,立体和对映选择性的广泛理论研究。呋喃-2-基甲氧基]苯基)丙烯酸酯衍生物(R1)和叠氮化物(R3和R3')以及硝酮(R2)用于类固醇的形成。在的反应R1和叠氮化物衍生物R3和R3' ,分子内(4 + 2)环加成R1是所述速率确定步骤(RDS)。还发现该反应是非常选择性的。发现吸电子基团取代的R1与R3的反应通常会增加rds的势垒,而溴除外,而发现给电子基团通常会降低rds的活化能。随后,我们报告了R1与环硝酮(R2),这比叠氮化物反应优越。全局反应性描述符的结果与激活势垒趋势高度吻合。发现该研究中所有考虑的反应都是动力学驱动的。
更新日期:2020-01-24
中文翻译:
串联分子内(4 + 2)/分子间(3 + 2)环加成反应形成聚氮杂和聚异恶唑烷类固醇的机理研究
通过有效和选择性的分子设计和合成来构建在生物医学中具有潜在应用潜力的分子的能力取决于对潜在反应机制的透彻了解。类固醇和相关杂环化合物的生物学重要性是众所周知的,但是缺乏旨在描述反应机理以补充实验者的努力的理论研究。本文中,我们报道了有关(E)-3-(2-[[]]的串联顺序分子内(4 + 2)/分子间(3 + 2)环加成反应的区域,立体和对映选择性的广泛理论研究。呋喃-2-基甲氧基]苯基)丙烯酸酯衍生物(R1)和叠氮化物(R3和R3')以及硝酮(R2)用于类固醇的形成。在的反应R1和叠氮化物衍生物R3和R3' ,分子内(4 + 2)环加成R1是所述速率确定步骤(RDS)。还发现该反应是非常选择性的。发现吸电子基团取代的R1与R3的反应通常会增加rds的势垒,而溴除外,而发现给电子基团通常会降低rds的活化能。随后,我们报告了R1与环硝酮(R2),这比叠氮化物反应优越。全局反应性描述符的结果与激活势垒趋势高度吻合。发现该研究中所有考虑的反应都是动力学驱动的。


























 京公网安备 11010802027423号
京公网安备 11010802027423号