当前位置:
X-MOL 学术
›
Vib. Spectrosc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
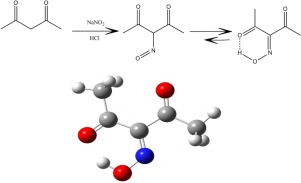
Synthesis, structure, tautomerism, intramolecular hydrogen bond, and vibrational assignment of 3-nitroso-2,4-pentanedione: A theoretical and experimental approach
Vibrational Spectroscopy ( IF 2.5 ) Pub Date : 2020-03-01 , DOI: 10.1016/j.vibspec.2020.103036 Mansoureh Rakhshanipour , Homa Jalali , Vahidreza Darugar , Hossein Eshghi , Mohammad Vakili
Vibrational Spectroscopy ( IF 2.5 ) Pub Date : 2020-03-01 , DOI: 10.1016/j.vibspec.2020.103036 Mansoureh Rakhshanipour , Homa Jalali , Vahidreza Darugar , Hossein Eshghi , Mohammad Vakili
|
Abstract The synthesis and molecular structure of 3-nitroso-2,4-pentanedione, entitled to oxime-acetylacetone (oxime-AA), was studied utilizing Density Functional Theory (DFT) calculations and the results were compared with those of 3-nitro-pentane-2,4-dione (NO2AA) and 2-nitromalonaldehyde (NO2MA). The vibrational frequencies of the most stable cis-enol form were calculated using the B3LYP functional and 6-311++G(d,p) basis set. The calculated frequencies and chemical shifts of oxime-AA were compared with the experimental results. The calculated geometrical parameters for oxime-AA show a medium hydrogen bond compared with their α-substituted (NO2AA) and (NO2MA) which manifested the strong hydrogen bond. The calculated O⋯O distance of 2.460–2.561 A is about 0.002-0.104 A longer than in NO2AA and NO2MA. According to the theoretical calculations, oxime-AA has a structure with a hydrogen bond strength of about 21.0 kcal/mol (calculated with 6-311++G(d,p) basis set), which is 4.0–6.0 kcal/mol weaker than the hydrogen bond strength of NO2AA and NO2MA. This decrease in the hydrogen bond strength is also consistent with the experimental results. Natural Bond Orbital (NBO) and AIM analyses were applied for considering the hydrogen bond strength in oxime-AA that indicates the effect of NOH group decreases the hydrogen bond strength.
中文翻译:

3-亚硝基-2,4-戊二酮的合成、结构、互变异构、分子内氢键和振动分配:一种理论和实验方法
摘要 利用密度泛函理论(DFT)计算研究了3-亚硝基-2,4-戊二酮(肟-乙酰丙酮(oxime-AA))的合成和分子结构,并将结果与3-硝基-乙酰丙酮(oxime-AA)的合成和分子结构进行了比较。戊烷-2,4-二酮 (NO2AA) 和 2-硝基丙二醛 (NO2MA)。使用 B3LYP 泛函和 6-311++G(d,p) 基组计算最稳定的顺式烯醇形式的振动频率。将计算的肟-AA 的频率和化学位移与实验结果进行比较。计算出的肟-AA 几何参数与其α-取代的(NO2AA) 和(NO2MA) 相比显示出中等氢键,后者表现出强氢键。计算出的 O⋯O 距离为 2.460-2.561 A,比 NO2AA 和 NO2MA 长约 0.002-0.104 A。根据理论计算,肟-AA具有氢键强度约为21.0 kcal/mol的结构(以6-311++G(d,p)基组计算),比其氢键强度弱4.0-6.0 kcal/mol NO2AA 和 NO2MA。氢键强度的这种降低也与实验结果一致。应用自然键轨道 (NBO) 和 AIM 分析来考虑肟-AA 中的氢键强度,表明 NOH 基团的影响会降低氢键强度。
更新日期:2020-03-01
中文翻译:
3-亚硝基-2,4-戊二酮的合成、结构、互变异构、分子内氢键和振动分配:一种理论和实验方法
摘要 利用密度泛函理论(DFT)计算研究了3-亚硝基-2,4-戊二酮(肟-乙酰丙酮(oxime-AA))的合成和分子结构,并将结果与3-硝基-乙酰丙酮(oxime-AA)的合成和分子结构进行了比较。戊烷-2,4-二酮 (NO2AA) 和 2-硝基丙二醛 (NO2MA)。使用 B3LYP 泛函和 6-311++G(d,p) 基组计算最稳定的顺式烯醇形式的振动频率。将计算的肟-AA 的频率和化学位移与实验结果进行比较。计算出的肟-AA 几何参数与其α-取代的(NO2AA) 和(NO2MA) 相比显示出中等氢键,后者表现出强氢键。计算出的 O⋯O 距离为 2.460-2.561 A,比 NO2AA 和 NO2MA 长约 0.002-0.104 A。根据理论计算,肟-AA具有氢键强度约为21.0 kcal/mol的结构(以6-311++G(d,p)基组计算),比其氢键强度弱4.0-6.0 kcal/mol NO2AA 和 NO2MA。氢键强度的这种降低也与实验结果一致。应用自然键轨道 (NBO) 和 AIM 分析来考虑肟-AA 中的氢键强度,表明 NOH 基团的影响会降低氢键强度。


























 京公网安备 11010802027423号
京公网安备 11010802027423号