Communications Physics ( IF 5.5 ) Pub Date : 2020-01-23 , DOI: 10.1038/s42005-020-0285-7 Akifumi Yamamura , Hiromasa Fujii , Hirohito Ogasawara , Dennis Nordlund , Osamu Takahashi , Yuutaro Kishi , Hiroyuki Ishii , Nobuhiko Kobayashi , Naoyuki Niitsu , Balthasar Blülle , Toshihiro Okamoto , Yusuke Wakabayashi , Shun Watanabe , Jun Takeya
|
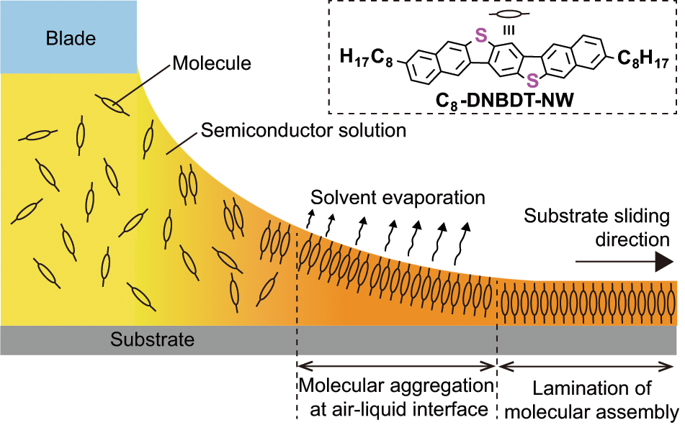
Arranging molecules into highly symmetric, topological crystal structures has been recognized as the best approach to functionalize electronic properties in molecular crystals, where the constituent molecules have been assumed to be rigid in shape. Here, in striking contrast, we demonstrate that the molecules in a monolayer organic crystal can undergo a significant deformation in proximity to the substrate, which is reflected by an asymmetry in the electron density profile. X-ray reflectivity and X-ray absorption spectroscopies in conjunction with density-functional theory calculations reveal that the highly planarized π-core are deformed into a bent shape, while the bulk lattice parameters are maintained. The molecular shape change is found to be perfectly suppressed in a bilayer single crystal, which leads to a 40% increase in mobility in the bilayer crystal. Our finding of a unique, sub-molecular scale shape change in monolayer single crystals can offer possibilities for functionalizing electrical properties via nano-scale physisorption.
中文翻译:
与单层有机单晶半导体的物理吸附界面处的亚分子结构弛豫
将分子排列成高度对称的拓扑晶体结构已被公认为是功能化分子晶体中电子特性的最佳方法,其中已假定构成分子的形状是刚性的。在这里,形成鲜明对比的是,我们证明了单层有机晶体中的分子在靠近基板的位置会发生明显的变形,这是通过电子密度分布图中的不对称性反映出来的。X射线反射率和X射线吸收光谱结合密度泛函理论计算表明,高度平坦的π核心变形为弯曲形状,同时保持整体晶格参数。发现在双层单晶体中分子形状的变化被完全抑制,这导致双层晶体中的迁移率增加了40%。我们在单层单晶中发现的独特的,亚分子尺度的形状变化,为通过纳米尺度的物理吸附功能化电性能提供了可能性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号