当前位置:
X-MOL 学术
›
J. Hum. Genet.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Novel NGLY1 gene variants in Chinese children with global developmental delay, microcephaly, hypotonia, hypertransaminasemia, alacrimia, and feeding difficulty.
Journal of Human Genetics ( IF 3.5 ) Pub Date : 2020-01-21 , DOI: 10.1038/s10038-019-0719-9 Kuerbanjiang Abuduxikuer 1 , Lin Zou 2 , Lei Wang 2 , Li Chen 2 , Jian-She Wang 1
Journal of Human Genetics ( IF 3.5 ) Pub Date : 2020-01-21 , DOI: 10.1038/s10038-019-0719-9 Kuerbanjiang Abuduxikuer 1 , Lin Zou 2 , Lei Wang 2 , Li Chen 2 , Jian-She Wang 1
Affiliation
|
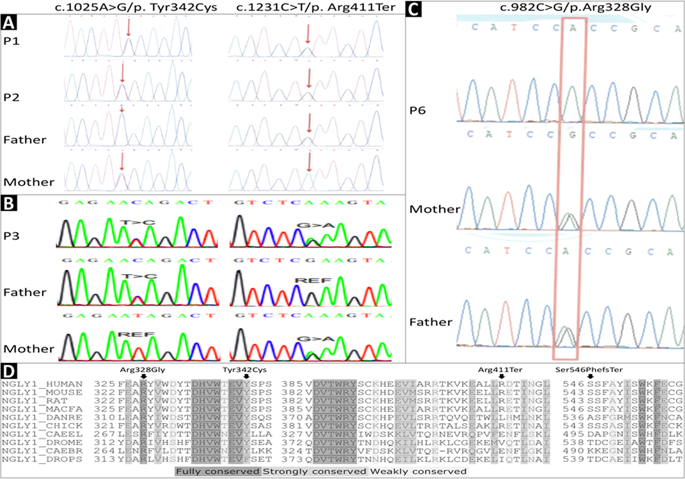
NGLY1 deficiency is the first and only autosomal recessive congenital disorder of N-linked deglycosylation (NGLY1-CDDG). To date, no patients with NGLY1 deficiency has been reported from mainland China or East Asia in English literature. Here, we present six patients with a diagnosis of NGLY1-CDDG on the basis of clinical phenotype, genetic testing, and functional studies. We retrospectively analyzed clinical phenotypes and NGLY1 genotypes of six cases from four families. Informed consent was obtained for diagnosis and treatment. In-silico tools and in vitro enzyme activity assays were used to determine pathogenicity of NGLY1 varaints. All patients had typical features of NGLY1-CDDG, including global developmental delay, microcephaly, hypotonia, hypertransaminasemia, alacrimia, and feeding difficulty. Dysmorphic features found in our patients include flat nasal bridge, loose and hollow cheeks, short stature, malnutrition, and ptosis. Pachylosis could be a novel cutaneous feature that may be explained by lack of sweat. We found three novel variants, including one missense (c.982C > G/p.Arg328Gly), one splice site (c.1003+3A > G), and one frame-shift (c.1637-1652delCATCTTTTGCTTATAT/p.Ser546PhefsTer) variant. All mutations were predicted to be disease causing with in-silico prediction tools, and affected at least one feature of gene splicing. Protein modeling showed missense variants may affect covalent bonding within the protein structure, or interrupt active/binding amino-acid residues. In vitro studies indicated that proteins carrying missense variants (p.Arg328Gly and p.Tyr342Cys) lost the enzyme activity. We expanded clinical phenotype and genetic mutation spectrum of NGLY1-CDDG by reporting six cases, three novel variants, and novel clinical features from mainland China.
更新日期:2020-01-21


























 京公网安备 11010802027423号
京公网安备 11010802027423号