当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Costless Performance Improvement in Machine Learning for Graph-Based Molecular Analysis.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-13 , DOI: 10.1021/acs.jcim.9b00816 Gyoung S Na 1 , Hyun Woo Kim 1 , Hyunju Chang 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-13 , DOI: 10.1021/acs.jcim.9b00816 Gyoung S Na 1 , Hyun Woo Kim 1 , Hyunju Chang 1
Affiliation
|
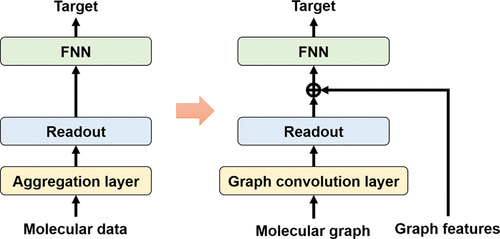
Graph neural networks (GNNs) have attracted significant attention from the chemical science community because molecules can be represented as a featured graph. In particular, graph convolutional network (GCN) and its variants have been widely used and have shown a state-of-the-art performance in analyzing molecules, such as molecular label classification, drug discovery, and molecular property prediction. However, in molecular analysis, existing GCNs have two fundamental limitations: (1) information of the molecular scale is distorted and (2) global structures in a molecule are ignored. These limitations can seriously degrade the performance in the machine learning-based molecular analysis because the scale and global structure information of a molecule occasionally have a significant effect on the molecular properties. To overcome the limitations of existing GCNs, we comprehensively analyzed the structure of GCNs and developed a costless solution for the limitations of GCNs. To demonstrate the effectiveness of our solution, extensive experiments were conducted on various benchmark datasets.
中文翻译:

基于图的分子分析的机器学习中无成本的性能改进。
图神经网络(GNN)引起了化学科学界的极大关注,因为分子可以表示为特征图。特别地,图卷积网络(GCN)及其变体已被广泛使用,并且在分析分子方面表现出了最先进的性能,例如分子标记分类,药物发现和分子特性预测。但是,在分子分析中,现有的GCN具有两个基本局限性:(1)分子尺度的信息失真,并且(2)分子中的整体结构被忽略。这些限制会严重降低基于机器学习的分子分析的性能,因为分子的规模和整体结构信息有时会对分子特性产生重大影响。为了克服现有GCN的局限性,我们全面分析了GCN的结构,并为GCN的局限性开发了一种无成本的解决方案。为了证明我们解决方案的有效性,我们对各种基准数据集进行了广泛的实验。
更新日期:2020-01-13
中文翻译:
基于图的分子分析的机器学习中无成本的性能改进。
图神经网络(GNN)引起了化学科学界的极大关注,因为分子可以表示为特征图。特别地,图卷积网络(GCN)及其变体已被广泛使用,并且在分析分子方面表现出了最先进的性能,例如分子标记分类,药物发现和分子特性预测。但是,在分子分析中,现有的GCN具有两个基本局限性:(1)分子尺度的信息失真,并且(2)分子中的整体结构被忽略。这些限制会严重降低基于机器学习的分子分析的性能,因为分子的规模和整体结构信息有时会对分子特性产生重大影响。为了克服现有GCN的局限性,我们全面分析了GCN的结构,并为GCN的局限性开发了一种无成本的解决方案。为了证明我们解决方案的有效性,我们对各种基准数据集进行了广泛的实验。

























 京公网安备 11010802027423号
京公网安备 11010802027423号