当前位置:
X-MOL 学术
›
Adv. Energy Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Impact of Noncovalent Sulfur–Fluorine Interaction Position on Properties, Structures, and Photovoltaic Performance in Naphthobisthiadiazole‐Based Semiconducting Polymers
Advanced Energy Materials ( IF 27.8 ) Pub Date : 2020-01-12 , DOI: 10.1002/aenm.201903278 Masahiko Saito 1 , Tomohiro Fukuhara 2 , Satoshi Kamimura 1 , Hiroyuki Ichikawa 3 , Hiroyuki Yoshida 3, 4 , Tomoyuki Koganezawa 5 , Yutaka Ie 6 , Yasunari Tamai 2, 7 , Hyung Do Kim 2 , Hideo Ohkita 2 , Itaru Osaka 1
Advanced Energy Materials ( IF 27.8 ) Pub Date : 2020-01-12 , DOI: 10.1002/aenm.201903278 Masahiko Saito 1 , Tomohiro Fukuhara 2 , Satoshi Kamimura 1 , Hiroyuki Ichikawa 3 , Hiroyuki Yoshida 3, 4 , Tomoyuki Koganezawa 5 , Yutaka Ie 6 , Yasunari Tamai 2, 7 , Hyung Do Kim 2 , Hideo Ohkita 2 , Itaru Osaka 1
Affiliation
|
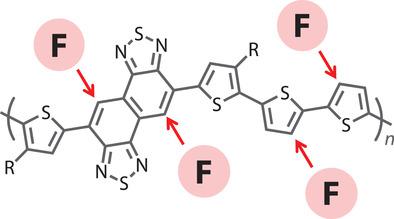
Controlling the energetics and backbone order of semiconducting polymers is essential for the performance improvement of polymer‐based solar cells. The use of fluorine as the substituent for the backbone is known to effectively deepen the molecular orbital energy levels and coplanarize the backbone by noncovalent interactions with sulfur of the thiophene ring. In this work, novel semiconducting polymers are designed and synthesized based on difluoronaphthobisthiadiazole (FNTz) as a new family of naphthobisthiadiazole (NTz)–quaterthiophene copolymer systems, which are one of the highest performing polymers in solar cells. The effect of the fluorination position on the energetics and backbone order is systematically studied. It is found that the dependence of the solar cell fill factor on the active layer thickness is very sensitive to the fluorination position. It is thus further investigated and discussed how the structural features of the polymers influence the photovoltaic parameters as well as the diode characteristics and bimolecular recombination. Further, the polymer with fluorine on both the naphthobisthiadiazole and quaterthiophene moieties exhibits a quite high power conversion efficiency of 10.8% in solar cells in combination with a fullerene. It is believed that the results would offer new insights into the development of semiconducting polymers.
中文翻译:

非共价硫-氟相互作用位置对萘二噻二唑基半导体聚合物的性质,结构和光伏性能的影响
控制半导体聚合物的能量和主链顺序对于提高基于聚合物的太阳能电池的性能至关重要。已知使用氟作为骨架的取代基可有效加深分子轨道能级,并通过与噻吩环的硫的非共价相互作用使骨架共平面化。在这项工作中,基于二氟萘二噻二唑(FNTz)作为萘二噻二唑(NTz)-四噻吩共聚物系统的新家族,设计并合成了新型半导体聚合物,这是太阳能电池中性能最高的聚合物之一。系统地研究了氟化位置对能量和主链顺序的影响。发现太阳能电池填充因子对有源层厚度的依赖性对氟化位置非常敏感。因此,将进一步研究和讨论聚合物的结构特征如何影响光伏参数以及二极管特性和双分子重组。此外,在萘二噻二唑和季噻吩部分上都具有氟的聚合物在与富勒烯组合的太阳能电池中表现出相当高的10.8%的功率转换效率。相信该结果将为半导体聚合物的开发提供新的见解。此外,在萘二噻二唑和季噻吩部分上都具有氟的聚合物在与富勒烯组合的太阳能电池中表现出相当高的10.8%的功率转换效率。相信该结果将为半导体聚合物的开发提供新的见解。此外,在萘二噻二唑和季噻吩部分上都具有氟的聚合物在与富勒烯组合的太阳能电池中表现出相当高的10.8%的功率转换效率。相信该结果将为半导体聚合物的开发提供新的见解。
更新日期:2020-02-18
中文翻译:
非共价硫-氟相互作用位置对萘二噻二唑基半导体聚合物的性质,结构和光伏性能的影响
控制半导体聚合物的能量和主链顺序对于提高基于聚合物的太阳能电池的性能至关重要。已知使用氟作为骨架的取代基可有效加深分子轨道能级,并通过与噻吩环的硫的非共价相互作用使骨架共平面化。在这项工作中,基于二氟萘二噻二唑(FNTz)作为萘二噻二唑(NTz)-四噻吩共聚物系统的新家族,设计并合成了新型半导体聚合物,这是太阳能电池中性能最高的聚合物之一。系统地研究了氟化位置对能量和主链顺序的影响。发现太阳能电池填充因子对有源层厚度的依赖性对氟化位置非常敏感。因此,将进一步研究和讨论聚合物的结构特征如何影响光伏参数以及二极管特性和双分子重组。此外,在萘二噻二唑和季噻吩部分上都具有氟的聚合物在与富勒烯组合的太阳能电池中表现出相当高的10.8%的功率转换效率。相信该结果将为半导体聚合物的开发提供新的见解。此外,在萘二噻二唑和季噻吩部分上都具有氟的聚合物在与富勒烯组合的太阳能电池中表现出相当高的10.8%的功率转换效率。相信该结果将为半导体聚合物的开发提供新的见解。此外,在萘二噻二唑和季噻吩部分上都具有氟的聚合物在与富勒烯组合的太阳能电池中表现出相当高的10.8%的功率转换效率。相信该结果将为半导体聚合物的开发提供新的见解。


























 京公网安备 11010802027423号
京公网安备 11010802027423号