当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Highly Accurate Prediction of Core Spectra of Molecules at Density Functional Theory Cost: Attaining Sub-electronvolt Error from a Restricted Open-Shell Kohn-Sham Approach.
The Journal of Physical Chemistry Letters ( IF 5.7 ) Pub Date : 2020-01-17 , DOI: 10.1021/acs.jpclett.9b03661 Diptarka Hait 1, 2 , Martin Head-Gordon 1, 2
The Journal of Physical Chemistry Letters ( IF 5.7 ) Pub Date : 2020-01-17 , DOI: 10.1021/acs.jpclett.9b03661 Diptarka Hait 1, 2 , Martin Head-Gordon 1, 2
Affiliation
|
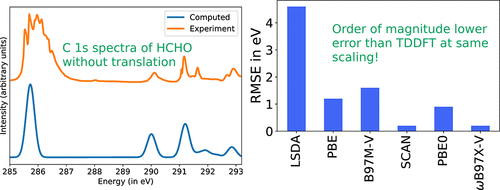
We present the use of the recently developed square gradient minimization (SGM) algorithm for excited-state orbital optimization to obtain spin-pure restricted open-shell Kohn-Sham (ROKS) energies for core excited states of molecules. The SGM algorithm is robust against variational collapse and offers a reliable route to converging orbitals for target excited states at only 2-3 times the cost of ground-state orbital optimization (per iteration). ROKS/SGM with the modern SCAN/ωB97X-V functionals is found to predict the K-edge of C, N, O, and F to a root mean squared error of ∼0.3 eV. ROKS/SGM is equally effective at predicting L-edge spectra of third period elements, provided a perturbative spin-orbit correction is employed. This high accuracy can be contrasted with traditional time-dependent density functional theory (TDDFT), which typically has greater than 10 eV error and requires translation of computed spectra to align with experiment. ROKS is computationally affordable (having the same scaling as ground-state DFT and a slightly larger prefactor) and can be applied to geometry optimizations/ab initio molecular dynamics of core excited states, as well as condensed phase simulations. ROKS can also model doubly excited/ionized states with one broken electron pair, which are beyond the ability of linear response based methods.
中文翻译:

以密度泛函理论为代价的分子核心光谱的高精度预测:通过受限的开壳式Kohn-Sham方法获得亚电子误差。
我们目前使用最新开发的平方梯度最小化(SGM)算法进行激发态轨道优化,以获得分子核心激发态的自旋纯受限开壳Kohn-Sham(ROKS)能量。SGM算法对变分崩溃具有鲁棒性,并为目标激发态的收敛轨道提供了可靠的途径,其成本仅为基态轨道优化(每次迭代)的2-3倍。具有现代SCAN /ωB97X-V功能的ROKS / SGM被发现可预测C,N,O和F的K边缘,均方根误差约为0.3 eV。如果采用微扰自旋轨道校正,ROKS / SGM在预测第三周期元素的L边缘谱方面同样有效。这种高准确性可以与传统的时变密度泛函理论(TDDFT)进行对比,通常具有大于10 eV的误差,并且需要转换计算光谱以与实验保持一致。ROKS的计算价格合理(具有与基态DFT相同的缩放比例,并且前置因子稍大),并且可以应用于几何优化/核心激发态的从头算分子动力学,以及凝聚态模拟。ROKS还可以用一个断裂的电子对对双激发/电离态进行建模,这超出了基于线性响应的方法的能力。
更新日期:2020-01-17
中文翻译:
以密度泛函理论为代价的分子核心光谱的高精度预测:通过受限的开壳式Kohn-Sham方法获得亚电子误差。
我们目前使用最新开发的平方梯度最小化(SGM)算法进行激发态轨道优化,以获得分子核心激发态的自旋纯受限开壳Kohn-Sham(ROKS)能量。SGM算法对变分崩溃具有鲁棒性,并为目标激发态的收敛轨道提供了可靠的途径,其成本仅为基态轨道优化(每次迭代)的2-3倍。具有现代SCAN /ωB97X-V功能的ROKS / SGM被发现可预测C,N,O和F的K边缘,均方根误差约为0.3 eV。如果采用微扰自旋轨道校正,ROKS / SGM在预测第三周期元素的L边缘谱方面同样有效。这种高准确性可以与传统的时变密度泛函理论(TDDFT)进行对比,通常具有大于10 eV的误差,并且需要转换计算光谱以与实验保持一致。ROKS的计算价格合理(具有与基态DFT相同的缩放比例,并且前置因子稍大),并且可以应用于几何优化/核心激发态的从头算分子动力学,以及凝聚态模拟。ROKS还可以用一个断裂的电子对对双激发/电离态进行建模,这超出了基于线性响应的方法的能力。


























 京公网安备 11010802027423号
京公网安备 11010802027423号