Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Synthesis and amide imidic prototropic tautomerization in thiophene-2-carbohydrazide: XRD, DFT/HSA-computation, DNA-docking, TG and isoconversional kinetics via FWO and KAS models
RSC Advances ( IF 3.9 ) Pub Date : 2020-1-10 , DOI: 10.1039/c9ra09831c Nabil Al-Zaqri 1 , Tamer Khatib 2 , Ali Alsalme 1 , Fahad A Alharthi 1 , Abdelkader Zarrouk 3 , Ismail Warad 4, 5
RSC Advances ( IF 3.9 ) Pub Date : 2020-1-10 , DOI: 10.1039/c9ra09831c Nabil Al-Zaqri 1 , Tamer Khatib 2 , Ali Alsalme 1 , Fahad A Alharthi 1 , Abdelkader Zarrouk 3 , Ismail Warad 4, 5
Affiliation
|
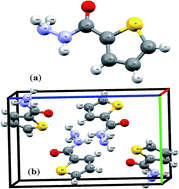
Thiophene-2-carbohydrazide as a novel small-molecule amide tautomer has been synthesized with an acceptable yield under microwave radiation (MW) conditions. The amide  imidic thiophene-2-carbohydrazide prototropic tautomerization via single proton intramigration was computed using the DFT B3LYP/6-311G(d,p) level of theory. The endo-isomer amide structure of thiophene-2-carbohydrazide was proved by XRD and is considered to be the kinetically favored isomer. The DFT-structure parameters were compared to their corresponding XRD-experimental parameters. Several H-bond interactions were detected in the crystal lattice experimentally using the XRD-packing model then correlated to MEP and HSA calculations. The manual and calculated electronic parameters such as, frontier molecular orbital energies, excitation energy, absorption, dipole moment, DOS, GRD quantum parameters and TD-SCF/B3LYP were DFT computed. The thiophene-2-carbohydrazide isomers together with their prototropic (E)/(Z)-thiophene-2-carbohydrazonic acid tautomers were docked against 1BNA DNA. FWO and KAS isoconversional kinetic methods were applied, and the thermal behavior and estimated Ea–α relations were determined.
imidic thiophene-2-carbohydrazide prototropic tautomerization via single proton intramigration was computed using the DFT B3LYP/6-311G(d,p) level of theory. The endo-isomer amide structure of thiophene-2-carbohydrazide was proved by XRD and is considered to be the kinetically favored isomer. The DFT-structure parameters were compared to their corresponding XRD-experimental parameters. Several H-bond interactions were detected in the crystal lattice experimentally using the XRD-packing model then correlated to MEP and HSA calculations. The manual and calculated electronic parameters such as, frontier molecular orbital energies, excitation energy, absorption, dipole moment, DOS, GRD quantum parameters and TD-SCF/B3LYP were DFT computed. The thiophene-2-carbohydrazide isomers together with their prototropic (E)/(Z)-thiophene-2-carbohydrazonic acid tautomers were docked against 1BNA DNA. FWO and KAS isoconversional kinetic methods were applied, and the thermal behavior and estimated Ea–α relations were determined.
中文翻译:

噻吩-2-碳酰肼中的合成和酰胺亚氨基质子互变异构化:XRD、DFT/HSA 计算、DNA 对接、TG 和通过 FWO 和 KAS 模型的等转化动力学
Thiophene-2-carbohydrazide 作为一种新型的小分子酰胺互变异构体已在微波辐射 (MW) 条件下以可接受的产率合成。使用 DFT B3LYP/6-311G(d,p) 水平的理论计算了通过单质子内迁移的酰胺亚氨基噻吩-2-碳酰肼质子互变异构化。远藤_XRD证明了噻吩-2-碳酰肼的-异构体酰胺结构,被认为是动力学有利的异构体。将 DFT 结构参数与其相应的 XRD 实验参数进行比较。使用 XRD 堆积模型在晶格中检测到几种 H 键相互作用,然后与 MEP 和 HSA 计算相关联。手动和计算的电子参数,如前沿分子轨道能量、激发能量、吸收、偶极矩、DOS、GRD量子参数和TD-SCF/B3LYP进行了DFT计算。噻吩-2-碳酰肼同分异构体及其质子 ( E )/( Z)-thiophene-2-carbohydrazonic acid 互变异构体与 1BNA DNA 对接。应用 FWO 和 KAS 等转化动力学方法,确定了热行为和估计的E a - α关系。
更新日期:2020-01-10
imidic thiophene-2-carbohydrazide prototropic tautomerization via single proton intramigration was computed using the DFT B3LYP/6-311G(d,p) level of theory. The endo-isomer amide structure of thiophene-2-carbohydrazide was proved by XRD and is considered to be the kinetically favored isomer. The DFT-structure parameters were compared to their corresponding XRD-experimental parameters. Several H-bond interactions were detected in the crystal lattice experimentally using the XRD-packing model then correlated to MEP and HSA calculations. The manual and calculated electronic parameters such as, frontier molecular orbital energies, excitation energy, absorption, dipole moment, DOS, GRD quantum parameters and TD-SCF/B3LYP were DFT computed. The thiophene-2-carbohydrazide isomers together with their prototropic (E)/(Z)-thiophene-2-carbohydrazonic acid tautomers were docked against 1BNA DNA. FWO and KAS isoconversional kinetic methods were applied, and the thermal behavior and estimated Ea–α relations were determined.
中文翻译:
噻吩-2-碳酰肼中的合成和酰胺亚氨基质子互变异构化:XRD、DFT/HSA 计算、DNA 对接、TG 和通过 FWO 和 KAS 模型的等转化动力学
Thiophene-2-carbohydrazide 作为一种新型的小分子酰胺互变异构体已在微波辐射 (MW) 条件下以可接受的产率合成。使用 DFT B3LYP/6-311G(d,p) 水平的理论计算了通过单质子内迁移的酰胺亚氨基
噻吩-2-碳酰肼质子互变异构化。远藤_XRD证明了噻吩-2-碳酰肼的-异构体酰胺结构,被认为是动力学有利的异构体。将 DFT 结构参数与其相应的 XRD 实验参数进行比较。使用 XRD 堆积模型在晶格中检测到几种 H 键相互作用,然后与 MEP 和 HSA 计算相关联。手动和计算的电子参数,如前沿分子轨道能量、激发能量、吸收、偶极矩、DOS、GRD量子参数和TD-SCF/B3LYP进行了DFT计算。噻吩-2-碳酰肼同分异构体及其质子 ( E )/( Z)-thiophene-2-carbohydrazonic acid 互变异构体与 1BNA DNA 对接。应用 FWO 和 KAS 等转化动力学方法,确定了热行为和估计的E a - α关系。


























 京公网安备 11010802027423号
京公网安备 11010802027423号