当前位置:
X-MOL 学术
›
Diam. Relat. Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
First principles study on the stability and thermodynamic properties of N Au co-doped graphene
Diamond and Related Materials ( IF 4.1 ) Pub Date : 2020-03-01 , DOI: 10.1016/j.diamond.2020.107704 Jianning Zhang , Ling Ma , Min Zhang , Jianmin Zhang
Diamond and Related Materials ( IF 4.1 ) Pub Date : 2020-03-01 , DOI: 10.1016/j.diamond.2020.107704 Jianning Zhang , Ling Ma , Min Zhang , Jianmin Zhang
|
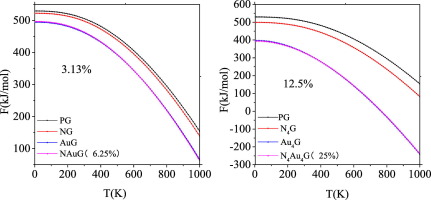
Abstract Based on first-principles density functional perturbation theory and first-principles molecular dynamics (MD) method, the effects of N and Au dopants as well as doping concentration on the structure stability, thermal stability and thermodynamic properties of graphene were systemically studied. It is found that the formation energy, phonon dispersion curve and MD simulation analysis indicate that N-doped graphene is more stable than Au and N Au co-doped graphene at the ground state or room temperature (300 K). The graphene doped with N, Au and N Au pair at different doping concentration can significantly affect its thermodynamic properties. Regardless of low or high concentrations, the specific heat and entropy of the doped graphene system increase as temperature increases, whereas free energy decreases. The results provide a theoretical basis for regulating the thermal conductivity of graphene and for relevant developing device applications.
中文翻译:

N Au共掺杂石墨烯稳定性和热力学性质的第一性原理研究
摘要 基于第一性原理密度泛函微扰理论和第一性原理分子动力学(MD)方法,系统研究了N、Au掺杂剂以及掺杂浓度对石墨烯结构稳定性、热稳定性和热力学性能的影响。发现形成能、声子色散曲线和MD模拟分析表明,在基态或室温(300 K)下,N掺杂石墨烯比Au和N Au共掺杂石墨烯更稳定。掺杂不同掺杂浓度的 N、Au 和 N Au 对的石墨烯会显着影响其热力学性能。无论低浓度或高浓度,掺杂石墨烯系统的比热和熵随着温度升高而增加,而自由能降低。
更新日期:2020-03-01
中文翻译:
N Au共掺杂石墨烯稳定性和热力学性质的第一性原理研究
摘要 基于第一性原理密度泛函微扰理论和第一性原理分子动力学(MD)方法,系统研究了N、Au掺杂剂以及掺杂浓度对石墨烯结构稳定性、热稳定性和热力学性能的影响。发现形成能、声子色散曲线和MD模拟分析表明,在基态或室温(300 K)下,N掺杂石墨烯比Au和N Au共掺杂石墨烯更稳定。掺杂不同掺杂浓度的 N、Au 和 N Au 对的石墨烯会显着影响其热力学性能。无论低浓度或高浓度,掺杂石墨烯系统的比热和熵随着温度升高而增加,而自由能降低。

























 京公网安备 11010802027423号
京公网安备 11010802027423号