当前位置:
X-MOL 学术
›
React. Chem. Eng.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Elucidating the role of solvents in acid catalyzed dehydration of biorenewable hydroxy-lactones
Reaction Chemistry & Engineering ( IF 3.9 ) Pub Date : 2020-01-07 , DOI: 10.1039/c9re00261h Gourav Shrivastav 1, 2, 3, 4, 5 , Tuhin S. Khan 1, 2, 3, 4, 5 , Manish Agarwal 3, 4, 5, 6 , M. Ali Haider 1, 2, 3, 4, 5
Reaction Chemistry & Engineering ( IF 3.9 ) Pub Date : 2020-01-07 , DOI: 10.1039/c9re00261h Gourav Shrivastav 1, 2, 3, 4, 5 , Tuhin S. Khan 1, 2, 3, 4, 5 , Manish Agarwal 3, 4, 5, 6 , M. Ali Haider 1, 2, 3, 4, 5
Affiliation
|
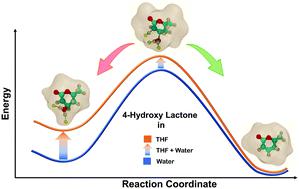
Owing to its low boiling point, tetrahydrofuran (THF) is an economical choice of solvent in biorenewables processing. Multifold acceleration in reaction rates is experimentally observed for Brønsted acid catalyzed reactions in THF, as compared to that in water. Herein, utilizing ab initio Car–Parrinello molecular dynamics (CPMD), classical molecular dynamics (MD) and density functional theory (DFT) simulations, a systematic theoretical framework is presented to explain the significant differences in the reactivity of Brønsted acid protons in THF as compared to that in water. The probe reaction of choice is the dehydration of 4-hydroxy-6-methyl lactone (HML) obtained from a biomass-derived substrate. Classical MD simulations elucidate the hydrogen bonding networks formed around the hydroxyl group of the reactant HML in explicit solvation environments of water and THF. The activation free energy barrier for the water removal step is calculated using CPMD–metadynamics simulations. In THF, the free energy barrier is 107 kJ mol−1, which is observed to be lower by 26 kJ mol−1 as compared to that in water. This significant difference in activation free energies for the dehydration step explains the difference in reactivity. Static DFT simulations further elaborated the effect of the first solvation shell around the hydroxyl substituent on describing the activation barriers of the dehydration reaction. While the solvent environment in DFT simulations is kept implicit in nature, few explicit molecules of THF and water are allowed to interact with the hydroxyl group and β-carbon of HML. The activation energy for the dehydration of HML is calculated to be 103 kJ mol−1 in the pure water environment. Akin to the difference in free energy barriers obtained from CPMD calculations, the activation energy calculated from DFT is observed to be 25 kJ mol−1 lower in THF as compared to that in water.
中文翻译:

阐明溶剂在酸催化生物可再生羟基内酯脱水中的作用
由于其低沸点,四氢呋喃(THF)是生物可再生能源加工中溶剂的经济选择。与水中的布朗斯台德酸催化反应相比,实验观察到了布朗斯台德酸催化反应的反应速率加快了许多倍。在这里,利用从头开始Car–Parrinello分子动力学(CPMD),经典分子动力学(MD)和密度泛函理论(DFT)模拟,提出了系统的理论框架来解释布朗斯台德酸质子在THF中的反应性与在水中的反应之间的显着差异。选择的探针反应是从生物质衍生的底物获得的4-羟基-6-甲基内酯(HML)脱水。经典的MD模拟阐明了在水和THF的明显溶剂化环境中,反应物HML羟基周围形成的氢键网络。脱水步骤的无活化能垒是使用CPMD-动力学模拟计算的。在THF中,自由能垒为107 kJ mol -1,观察到低了26 kJ mol -1与水中相比 脱水步骤中活化自由能的显着差异解释了反应性的差异。静态DFT模拟进一步阐述了羟基取代基周围的第一个溶剂化壳层对描述脱水反应活化障碍的影响。尽管DFT模拟中的溶剂环境本质上是隐性的,但几乎没有THF和水的显式分子与HML的羟基和β-碳相互作用。在纯水环境中,HML脱水的活化能经计算为103 kJ mol -1。类似于通过CPMD计算获得的自由能垒的差异,从DFT计算得出的活化能为25 kJ mol -1 与水相比,四氢呋喃的含量更低。
更新日期:2020-01-07
中文翻译:
阐明溶剂在酸催化生物可再生羟基内酯脱水中的作用
由于其低沸点,四氢呋喃(THF)是生物可再生能源加工中溶剂的经济选择。与水中的布朗斯台德酸催化反应相比,实验观察到了布朗斯台德酸催化反应的反应速率加快了许多倍。在这里,利用从头开始Car–Parrinello分子动力学(CPMD),经典分子动力学(MD)和密度泛函理论(DFT)模拟,提出了系统的理论框架来解释布朗斯台德酸质子在THF中的反应性与在水中的反应之间的显着差异。选择的探针反应是从生物质衍生的底物获得的4-羟基-6-甲基内酯(HML)脱水。经典的MD模拟阐明了在水和THF的明显溶剂化环境中,反应物HML羟基周围形成的氢键网络。脱水步骤的无活化能垒是使用CPMD-动力学模拟计算的。在THF中,自由能垒为107 kJ mol -1,观察到低了26 kJ mol -1与水中相比 脱水步骤中活化自由能的显着差异解释了反应性的差异。静态DFT模拟进一步阐述了羟基取代基周围的第一个溶剂化壳层对描述脱水反应活化障碍的影响。尽管DFT模拟中的溶剂环境本质上是隐性的,但几乎没有THF和水的显式分子与HML的羟基和β-碳相互作用。在纯水环境中,HML脱水的活化能经计算为103 kJ mol -1。类似于通过CPMD计算获得的自由能垒的差异,从DFT计算得出的活化能为25 kJ mol -1 与水相比,四氢呋喃的含量更低。

























 京公网安备 11010802027423号
京公网安备 11010802027423号