当前位置:
X-MOL 学术
›
J. Hum. Genet.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Genetic screening for potassium channel mutations in Japanese autosomal dominant spinocerebellar ataxia.
Journal of Human Genetics ( IF 3.5 ) Pub Date : 2020-01-07 , DOI: 10.1038/s10038-019-0717-y Yui Tada 1, 2 , Kodai Kume 1 , Yukiko Matsuda 1 , Takashi Kurashige 3 , Yuhei Kanaya 1 , Ryosuke Ohsawa 1 , Hiroyuki Morino 1 , Hayato Tabu 4 , Satoshi Kaneko 5 , Toshihiko Suenaga 6 , Akira Kakizuka 2 , Hideshi Kawakami 1
Journal of Human Genetics ( IF 3.5 ) Pub Date : 2020-01-07 , DOI: 10.1038/s10038-019-0717-y Yui Tada 1, 2 , Kodai Kume 1 , Yukiko Matsuda 1 , Takashi Kurashige 3 , Yuhei Kanaya 1 , Ryosuke Ohsawa 1 , Hiroyuki Morino 1 , Hayato Tabu 4 , Satoshi Kaneko 5 , Toshihiko Suenaga 6 , Akira Kakizuka 2 , Hideshi Kawakami 1
Affiliation
|
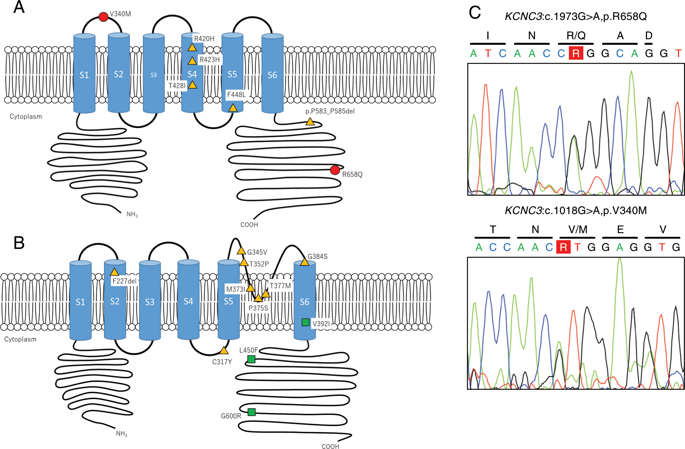
Spinocerebellar ataxia (SCA) is a genetically heterogeneous disease characterized by cerebellar ataxia. Many causative genes have been identified to date, the most common etiology being the abnormal expansion of repeat sequences, and the mutation of ion channel genes also play an important role in the development of SCA. Some of them encode calcium and potassium channels. However, due to limited reports about potassium genes in SCA, we screened 192 Japanese individuals with dominantly inherited SCA who had no abnormal repeat expansions of causative genes for potassium channel mutations (KCNC3 for SCA13 and KCND3 for SCA19/SCA22) by target sequencing. As a result, two variants were identified from two patients: c.1973G>A, p.R658Q and c.1018G>A, p.V340M for KCNC3, and no pathogenic variant was identified for KCND3. The newly identified p.V340M exists in the extracellular domain, and p.R658Q exists in the intracellular domain on the C-terminal side, although most of the reported KCNC3 mutations are present at the transmembrane site. Adult-onset and slowly progressive cerebellar ataxia are the main clinical features of SCA13 and SCA19 caused by potassium channel mutations, which was similar in our cases. SCA13 caused by KCNC3 mutations may present with deep sensory loss and cognitive impairment in addition to cerebellar ataxia. In this study, mild deep sensory loss was observed in one case. SCA caused by potassium channel gene mutations is extremely rare, and more cases should be accumulated in the future to elucidate its pathogenesis due to channel dysfunction.
中文翻译:

日本常染色体显性遗传性脊髓小脑共济失调钾通道突变的遗传筛选。
脊髓小脑性共济失调(SCA)是一种以小脑性共济失调为特征的遗传异质性疾病。迄今为止,已经鉴定出许多致病基因,最常见的病因是重复序列的异常扩增,并且离子通道基因的突变在SCA的发展中也起着重要作用。其中一些编码钙和钾通道。但是,由于关于SCA中钾基因的报道有限,我们通过靶标测序筛选了192名遗传性SCA的日本人,这些人没有钾通道突变的致病基因(SCA13的KCNC3和SCA19 / SCA22的KCND3)的异常重复扩增。结果,从两名患者中鉴定出两个变体:KCNC3的c.1973G> A,p.R658Q和c.1018G> A,p.V340M,并且未鉴定出KCND3的致病性变体。新确定的p。V340M存在于细胞外结构域中,而p.R658Q存在于C端侧的细胞内结构域中,尽管大多数报道的KCNC3突变都存在于跨膜位点。成人发作和缓慢进行性小脑性共济失调是由钾通道突变引起的SCA13和SCA19的主要临床特征,与我们的病例相似。除小脑性共济失调外,由KCNC3突变引起的SCA13可能还伴有深度感觉丧失和认知障碍。在这项研究中,在一个案例中观察到了轻微的深度感觉丧失。由钾通道基因突变引起的SCA极为罕见,将来应积累更多病例,以阐明由于通道功能异常引起的发病机理。尽管大多数报道的KCNC3突变都存在于跨膜位点。成人发作和缓慢进行性小脑性共济失调是由钾通道突变引起的SCA13和SCA19的主要临床特征,与我们的病例相似。除小脑性共济失调外,由KCNC3突变引起的SCA13可能还伴有深度感觉丧失和认知障碍。在这项研究中,在一个案例中观察到了轻微的深度感觉丧失。由钾通道基因突变引起的SCA极为罕见,将来应积累更多病例,以阐明由于通道功能异常引起的发病机理。尽管大多数报道的KCNC3突变都存在于跨膜位点。成人发作和缓慢进行性小脑性共济失调是由钾通道突变引起的SCA13和SCA19的主要临床特征,与我们的病例相似。除小脑性共济失调外,由KCNC3突变引起的SCA13可能还伴有深度感觉丧失和认知障碍。在这项研究中,在一个案例中观察到了轻微的深度感觉丧失。由钾通道基因突变引起的SCA极为罕见,将来应积累更多病例,以阐明由于通道功能异常引起的发病机理。在我们的案例中是相似的。除小脑性共济失调外,由KCNC3突变引起的SCA13可能还伴有深度感觉丧失和认知障碍。在这项研究中,在一个案例中观察到了轻微的深度感觉丧失。由钾通道基因突变引起的SCA极为罕见,将来应积累更多病例,以阐明由于通道功能异常引起的发病机理。在我们的案例中是相似的。除小脑性共济失调外,由KCNC3突变引起的SCA13可能还伴有深度感觉丧失和认知障碍。在这项研究中,在一个案例中观察到了轻微的深度感觉丧失。由钾通道基因突变引起的SCA极为罕见,将来应积累更多病例,以阐明由于通道功能异常引起的发病机理。
更新日期:2020-01-07
中文翻译:
日本常染色体显性遗传性脊髓小脑共济失调钾通道突变的遗传筛选。
脊髓小脑性共济失调(SCA)是一种以小脑性共济失调为特征的遗传异质性疾病。迄今为止,已经鉴定出许多致病基因,最常见的病因是重复序列的异常扩增,并且离子通道基因的突变在SCA的发展中也起着重要作用。其中一些编码钙和钾通道。但是,由于关于SCA中钾基因的报道有限,我们通过靶标测序筛选了192名遗传性SCA的日本人,这些人没有钾通道突变的致病基因(SCA13的KCNC3和SCA19 / SCA22的KCND3)的异常重复扩增。结果,从两名患者中鉴定出两个变体:KCNC3的c.1973G> A,p.R658Q和c.1018G> A,p.V340M,并且未鉴定出KCND3的致病性变体。新确定的p。V340M存在于细胞外结构域中,而p.R658Q存在于C端侧的细胞内结构域中,尽管大多数报道的KCNC3突变都存在于跨膜位点。成人发作和缓慢进行性小脑性共济失调是由钾通道突变引起的SCA13和SCA19的主要临床特征,与我们的病例相似。除小脑性共济失调外,由KCNC3突变引起的SCA13可能还伴有深度感觉丧失和认知障碍。在这项研究中,在一个案例中观察到了轻微的深度感觉丧失。由钾通道基因突变引起的SCA极为罕见,将来应积累更多病例,以阐明由于通道功能异常引起的发病机理。尽管大多数报道的KCNC3突变都存在于跨膜位点。成人发作和缓慢进行性小脑性共济失调是由钾通道突变引起的SCA13和SCA19的主要临床特征,与我们的病例相似。除小脑性共济失调外,由KCNC3突变引起的SCA13可能还伴有深度感觉丧失和认知障碍。在这项研究中,在一个案例中观察到了轻微的深度感觉丧失。由钾通道基因突变引起的SCA极为罕见,将来应积累更多病例,以阐明由于通道功能异常引起的发病机理。尽管大多数报道的KCNC3突变都存在于跨膜位点。成人发作和缓慢进行性小脑性共济失调是由钾通道突变引起的SCA13和SCA19的主要临床特征,与我们的病例相似。除小脑性共济失调外,由KCNC3突变引起的SCA13可能还伴有深度感觉丧失和认知障碍。在这项研究中,在一个案例中观察到了轻微的深度感觉丧失。由钾通道基因突变引起的SCA极为罕见,将来应积累更多病例,以阐明由于通道功能异常引起的发病机理。在我们的案例中是相似的。除小脑性共济失调外,由KCNC3突变引起的SCA13可能还伴有深度感觉丧失和认知障碍。在这项研究中,在一个案例中观察到了轻微的深度感觉丧失。由钾通道基因突变引起的SCA极为罕见,将来应积累更多病例,以阐明由于通道功能异常引起的发病机理。在我们的案例中是相似的。除小脑性共济失调外,由KCNC3突变引起的SCA13可能还伴有深度感觉丧失和认知障碍。在这项研究中,在一个案例中观察到了轻微的深度感觉丧失。由钾通道基因突变引起的SCA极为罕见,将来应积累更多病例,以阐明由于通道功能异常引起的发病机理。


























 京公网安备 11010802027423号
京公网安备 11010802027423号