当前位置:
X-MOL 学术
›
Mech. Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
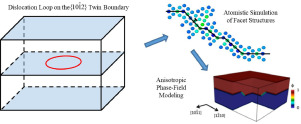
Structure and Kinetics of Three-Dimensional Defects on the {101¯2} Twin Boundary in Magnesium: Atomistic and Phase-field Simulations
Mechanics of Materials ( IF 3.9 ) Pub Date : 2020-04-01 , DOI: 10.1016/j.mechmat.2020.103314 Douglas E. Spearot , Vincent Taupin , Khanh Dang , Laurent Capolungo
Mechanics of Materials ( IF 3.9 ) Pub Date : 2020-04-01 , DOI: 10.1016/j.mechmat.2020.103314 Douglas E. Spearot , Vincent Taupin , Khanh Dang , Laurent Capolungo
|
Abstract Twinning is an important plastic deformation mechanism in Mg. While computational and experimental efforts have studied the two-dimensional equilibrium structure of boundaries and facets that surround twin domains, little consideration has been given to three-dimensional nonequilibrium structures. Furthermore, the relationship between the structure of nonequilibrium facets and the kinetics of twinning is also not well understood. Thus, the objective of this work is to characterize the structure of three-dimensional defects on the { 10 1 ¯ 2 } twin boundary and to study the kinetics associated with motion of the facets that bound these three-dimensional defects. Atomistic simulations show that the three-dimensional defects are bounded by nonequilibrium facets with prismatic/basal and twist pyramidal/pyramidal interfaces. The three-dimensional defects are surrounded on the { 10 1 ¯ 2 } twin boundary by disconnection loops. The kinetics of the twin domain facets at finite temperature are analyzed by both molecular dynamics and a newly proposed anisotropic phase-field model. The latter allows the deconvolution of the competing role of interface energy, mobility and internal stress state. Molecular dynamics simulations show that inclined facets control the annihilation process; this behavior is captured in the phase-field model using mobility for the coherent twin boundary that is significantly lower than that of crater inclined facets. Furthermore, molecular dynamics simulation results are best matched via the introduction of facet orientation dependent excess energies.
中文翻译:

镁中{101¯2}孪晶界三维缺陷的结构和动力学:原子和相场模拟
摘要 孪晶是镁合金中一种重要的塑性变形机制。虽然计算和实验工作研究了围绕双域的边界和小平面的二维平衡结构,但很少考虑三维非平衡结构。此外,非平衡面的结构与孪晶动力学之间的关系也不是很清楚。因此,这项工作的目的是表征 { 10 1 ¯ 2 } 双晶边界上三维缺陷的结构,并研究与约束这些三维缺陷的小平面运动相关的动力学。原子模拟表明,三维缺陷由具有棱柱/基底和扭曲棱锥/棱锥界面的非平衡小平面限定。三维缺陷在 { 10 1 ¯ 2 } 孪晶边界上被断开环包围。通过分子动力学和新提出的各向异性相场模型分析了有限温度下双畴面的动力学。后者允许对界面能、迁移率和内应力状态的竞争作用进行解卷积。分子动力学模拟表明倾斜面控制着湮灭过程;这种行为在相场模型中被捕获,使用相干孪晶边界的迁移率明显低于陨石坑倾斜面的迁移率。此外,分子动力学模拟结果通过引入依赖于小平面取向的过剩能量而得到最佳匹配。通过分子动力学和新提出的各向异性相场模型分析了有限温度下双畴面的动力学。后者允许对界面能、迁移率和内应力状态的竞争作用进行解卷积。分子动力学模拟表明倾斜面控制着湮灭过程;这种行为在相场模型中被捕获,使用相干孪晶边界的迁移率明显低于陨石坑倾斜面的迁移率。此外,分子动力学模拟结果通过引入依赖于小平面取向的过剩能量而得到最佳匹配。通过分子动力学和新提出的各向异性相场模型分析了有限温度下双畴面的动力学。后者允许对界面能、迁移率和内应力状态的竞争作用进行解卷积。分子动力学模拟表明倾斜面控制着湮灭过程;这种行为在相场模型中被捕获,使用相干孪晶边界的迁移率明显低于陨石坑倾斜面的迁移率。此外,分子动力学模拟结果通过引入依赖于小平面取向的过剩能量而得到最佳匹配。后者允许对界面能、迁移率和内应力状态的竞争作用进行解卷积。分子动力学模拟表明倾斜面控制着湮灭过程;这种行为在相场模型中被捕获,使用相干孪晶边界的迁移率明显低于陨石坑倾斜面的迁移率。此外,分子动力学模拟结果通过引入依赖于小平面取向的过剩能量而得到最佳匹配。后者允许对界面能、迁移率和内部应力状态的竞争作用进行解卷积。分子动力学模拟表明倾斜面控制着湮灭过程;这种行为在相场模型中被捕获,使用相干孪晶边界的迁移率明显低于陨石坑倾斜面的迁移率。此外,分子动力学模拟结果最好通过引入依赖于小平面取向的过剩能量来匹配。
更新日期:2020-04-01
中文翻译:
镁中{101¯2}孪晶界三维缺陷的结构和动力学:原子和相场模拟
摘要 孪晶是镁合金中一种重要的塑性变形机制。虽然计算和实验工作研究了围绕双域的边界和小平面的二维平衡结构,但很少考虑三维非平衡结构。此外,非平衡面的结构与孪晶动力学之间的关系也不是很清楚。因此,这项工作的目的是表征 { 10 1 ¯ 2 } 双晶边界上三维缺陷的结构,并研究与约束这些三维缺陷的小平面运动相关的动力学。原子模拟表明,三维缺陷由具有棱柱/基底和扭曲棱锥/棱锥界面的非平衡小平面限定。三维缺陷在 { 10 1 ¯ 2 } 孪晶边界上被断开环包围。通过分子动力学和新提出的各向异性相场模型分析了有限温度下双畴面的动力学。后者允许对界面能、迁移率和内应力状态的竞争作用进行解卷积。分子动力学模拟表明倾斜面控制着湮灭过程;这种行为在相场模型中被捕获,使用相干孪晶边界的迁移率明显低于陨石坑倾斜面的迁移率。此外,分子动力学模拟结果通过引入依赖于小平面取向的过剩能量而得到最佳匹配。通过分子动力学和新提出的各向异性相场模型分析了有限温度下双畴面的动力学。后者允许对界面能、迁移率和内应力状态的竞争作用进行解卷积。分子动力学模拟表明倾斜面控制着湮灭过程;这种行为在相场模型中被捕获,使用相干孪晶边界的迁移率明显低于陨石坑倾斜面的迁移率。此外,分子动力学模拟结果通过引入依赖于小平面取向的过剩能量而得到最佳匹配。通过分子动力学和新提出的各向异性相场模型分析了有限温度下双畴面的动力学。后者允许对界面能、迁移率和内应力状态的竞争作用进行解卷积。分子动力学模拟表明倾斜面控制着湮灭过程;这种行为在相场模型中被捕获,使用相干孪晶边界的迁移率明显低于陨石坑倾斜面的迁移率。此外,分子动力学模拟结果通过引入依赖于小平面取向的过剩能量而得到最佳匹配。后者允许对界面能、迁移率和内应力状态的竞争作用进行解卷积。分子动力学模拟表明倾斜面控制着湮灭过程;这种行为在相场模型中被捕获,使用相干孪晶边界的迁移率明显低于陨石坑倾斜面的迁移率。此外,分子动力学模拟结果通过引入依赖于小平面取向的过剩能量而得到最佳匹配。后者允许对界面能、迁移率和内部应力状态的竞争作用进行解卷积。分子动力学模拟表明倾斜面控制着湮灭过程;这种行为在相场模型中被捕获,使用相干孪晶边界的迁移率明显低于陨石坑倾斜面的迁移率。此外,分子动力学模拟结果最好通过引入依赖于小平面取向的过剩能量来匹配。


























 京公网安备 11010802027423号
京公网安备 11010802027423号