Computational and Theoretical Chemistry ( IF 2.8 ) Pub Date : 2020-01-07 , DOI: 10.1016/j.comptc.2020.112702 Seifollah Jalili , Mohammad Keshavarz
|
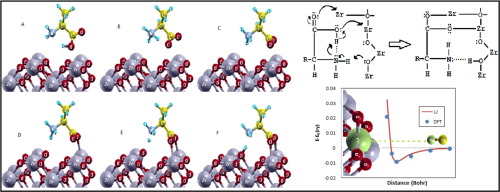
In order to determine the adsorption mechanism of molecules on zirconia and factors affecting it, the adsorption of a wide range of small molecules (He, H2, O2, N2, CO, NH3, H2O, H2CO), alkanes (CH4,C2H6, C3H8), and amino acids (glycine, alanine, proline, hydroxyproline, aspartic acid) were studied on the cubic ZrO2(110) surface by density functional theory method, along with the dispersion correction (DFT-D3). It was found that zirconium d orbitals, nonbonding electron pairs of the adsorbed atoms, and surface oxygen atoms play an important role in the adsorption mechanism. In addition, it was found that in the physical adsorption process, the interaction of atoms with the surface can be approximated by functional form of the 6-12 Lennard-Jones potential. Parameters and functional form of interaction potential help us establish a validated force field using high-level quantum mechanical calculations.
中文翻译:
氧化锆(110)表面吸附行为-密度泛函理论研究
为了确定分子在氧化锆上的吸附机理及其影响因素,对各种小分子(He,H 2,O 2,N 2,CO,NH 3,H 2 O,H 2 CO)进行吸附通过密度泛函理论方法研究了立方ZrO 2(110)表面上的烷烃(CH 4,C 2 H 6,C 3 H 8)和氨基酸(甘氨酸,丙氨酸,脯氨酸,羟脯氨酸,天冬氨酸),以及使用色散校正(DFT-D3)。发现锆d轨道,吸附原子的非键电子对和表面氧原子在吸附机理中起重要作用。此外,发现在物理吸附过程中,原子与表面的相互作用可以通过6-12 Lennard-Jones势的功能形式来近似。相互作用势的参数和功能形式可帮助我们使用高级量子力学计算来建立经过验证的力场。


























 京公网安备 11010802027423号
京公网安备 11010802027423号