当前位置:
X-MOL 学术
›
BBA Gen. Subj.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Identification and characterization of fragment binding sites for allosteric ligand design using the site identification by ligand competitive saturation hotspots approach (SILCS-Hotspots).
Biochimica et Biophysica Acta (BBA) - General Subjects ( IF 3 ) Pub Date : 2020-01-03 , DOI: 10.1016/j.bbagen.2020.129519 Alexander D MacKerell 1 , Sunhwan Jo 2 , Sirish Kaushik Lakkaraju 2 , Christoffer Lind 1 , Wenbo Yu 1
Biochimica et Biophysica Acta (BBA) - General Subjects ( IF 3 ) Pub Date : 2020-01-03 , DOI: 10.1016/j.bbagen.2020.129519 Alexander D MacKerell 1 , Sunhwan Jo 2 , Sirish Kaushik Lakkaraju 2 , Christoffer Lind 1 , Wenbo Yu 1
Affiliation
|
BACKGROUND
Fragment-based ligand design is used for the development of novel ligands that target macromolecules, most notably proteins. Central to its success is the identification of fragment binding sites that are spatially adjacent such that fragments occupying those sites may be linked to create drug-like ligands. Current experimental and computational approaches that address this problem typically identify only a limited number of sites as well as use a limited number of fragment types.
METHODS
The site-identification by ligand competitive saturation (SILCS) approach is extended to the identification of fragment bindings sites, with the method termed SILCS-Hotspots. The approach involves precomputation of the SILCS FragMaps following which the identification of Hotspots, performed by identifying of all possible fragment binding sites on the full 3D structure of the protein followed by spatial clustering.
RESULTS
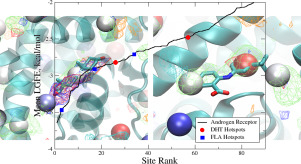
The SILCS-Hotspots approach identifies a large number of sites on the target protein, including many sites not accessible in experimental structures due to low binding affinities and binding sites on the protein interior. The identified sites are shown to recapitulate the location of known drug-like molecules in both allosteric and orthosteric binding sites on seven proteins including the androgen receptor, the CDK2 and Erk5 kinases, PTP1B phosphatase and three GPCRs; the β2-adrenergic, GPR40 fatty-acid binding and M2-muscarinic receptors. Analysis indicates the importance of considering all possible fragment binding sites, and not just those accessible to experimental methods, when identifying novel binding sites and performing ligand design versus just considering the most favorable sites. The approach is shown to identify a larger number of known binding sites of drug-like molecules versus the commonly used FTMap and Fpocket methods.
GENERAL SIGNIFICANCE
The present results indicate the potential utility of the SILCS-Hotspots approach for fragment-based rational design of ligands, including allosteric modulators.
中文翻译:

使用配体竞争性饱和热点方法(SILCS-Hotspots)进行位点识别,从而对变构配体设计中的片段结合位点进行识别和表征。
背景技术基于片段的配体设计用于开发靶向大分子,最特别是蛋白质的新型配体。其成功的关键是鉴定在空间上相邻的片段结合位点,使得占据那些位点的片段可以被连接以产生药物样配体。解决该问题的当前实验和计算方法通常仅识别有限数量的位点,并且使用有限数量的片段类型。方法通过配体竞争饱和(SILCS)方法进行的位点识别扩展到片段结合位点的识别,方法称为SILCS-Hotspots。该方法涉及对SILCS FragMaps进行预计算,然后识别热点,通过鉴定蛋白质完整3D结构上所有可能的片段结合位点,然后进行空间聚类来进行分析。结果SILCS-Hotspots方法可识别靶蛋白上的大量位点,包括由于结合亲和力和蛋白内部结合位点低而无法在实验结构中访问的许多位点。鉴定出的位点可概括已知药物样分子在七个蛋白(包括雄激素受体,CDK2和Erk5激酶,PTP1B磷酸酶和三个GPCR)的变构和正构结合位点中的位置。β2-肾上腺素,GPR40脂肪酸结合和M2-毒蕈碱受体。分析表明,考虑所有可能的片段结合位点,而不仅仅是实验方法可及的那些位点的重要性,在确定新的结合位点并进行配体设计时,只考虑最有利的位点。与常用的FTMap和Fpocket方法相比,该方法显示出可识别更多的类药物分子已知结合位点。一般意义本研究结果表明,SILCS-Hotspots方法可用于基于片段的配体(包括变构调节剂)的合理设计。
更新日期:2020-01-04
中文翻译:
使用配体竞争性饱和热点方法(SILCS-Hotspots)进行位点识别,从而对变构配体设计中的片段结合位点进行识别和表征。
背景技术基于片段的配体设计用于开发靶向大分子,最特别是蛋白质的新型配体。其成功的关键是鉴定在空间上相邻的片段结合位点,使得占据那些位点的片段可以被连接以产生药物样配体。解决该问题的当前实验和计算方法通常仅识别有限数量的位点,并且使用有限数量的片段类型。方法通过配体竞争饱和(SILCS)方法进行的位点识别扩展到片段结合位点的识别,方法称为SILCS-Hotspots。该方法涉及对SILCS FragMaps进行预计算,然后识别热点,通过鉴定蛋白质完整3D结构上所有可能的片段结合位点,然后进行空间聚类来进行分析。结果SILCS-Hotspots方法可识别靶蛋白上的大量位点,包括由于结合亲和力和蛋白内部结合位点低而无法在实验结构中访问的许多位点。鉴定出的位点可概括已知药物样分子在七个蛋白(包括雄激素受体,CDK2和Erk5激酶,PTP1B磷酸酶和三个GPCR)的变构和正构结合位点中的位置。β2-肾上腺素,GPR40脂肪酸结合和M2-毒蕈碱受体。分析表明,考虑所有可能的片段结合位点,而不仅仅是实验方法可及的那些位点的重要性,在确定新的结合位点并进行配体设计时,只考虑最有利的位点。与常用的FTMap和Fpocket方法相比,该方法显示出可识别更多的类药物分子已知结合位点。一般意义本研究结果表明,SILCS-Hotspots方法可用于基于片段的配体(包括变构调节剂)的合理设计。

























 京公网安备 11010802027423号
京公网安备 11010802027423号