当前位置:
X-MOL 学术
›
Bioorgan. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Synthesis and evaluation of novel S-benzyl- and S-alkylphthalimide- oxadiazole -benzenesulfonamide hybrids as inhibitors of dengue virus protease.
Bioorganic Chemistry ( IF 5.1 ) Pub Date : 2020-01-03 , DOI: 10.1016/j.bioorg.2020.103567 Syeda Shamila Hamdani 1 , Bilal Ahmad Khan 2 , Shahid Hameed 3 , Farwa Batool 4 , Hafiza Nosheen Saleem 4 , Ehsan Ullah Mughal 5 , Muhammad Saeed 4
Bioorganic Chemistry ( IF 5.1 ) Pub Date : 2020-01-03 , DOI: 10.1016/j.bioorg.2020.103567 Syeda Shamila Hamdani 1 , Bilal Ahmad Khan 2 , Shahid Hameed 3 , Farwa Batool 4 , Hafiza Nosheen Saleem 4 , Ehsan Ullah Mughal 5 , Muhammad Saeed 4
Affiliation
|
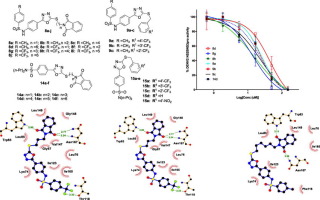
Direct acting antiviral drugs (DAADs) are becoming therapeutics of choice for the treatment of viral infections. Successful development of anti HIV and HCV drugs by targeting the viral proteases has provided impetus for discovering newer DAADs. Dengue virus (DENV) protease, which is composed of two nonstructural proteins, NS2B and NS3pro, can be likewise exploited for discovering new anti-dengue therapeutics. In this study, we have linked together two pharmaceutically interesting motifs, namely 1,3,4-oxadiazole and benzenesulfonamide in two alternative series to develop novel S-benzylated and S-alkylphthalimidated hybrids. For the first series of hybrids, 4-aminobenzoic acid (1) was reacted with substituted benzenesulfonyl chlorides via its amino group, whereas the carboxylic acid side was elaborated to sulfonamido-1,3,4-oxadiazole-2-thiols (6a/b) in three steps. At this stage, the intermediates 6a/b were bifurcated to either S-alkylphthalimidated (8a-j) or S-benzylated (9a-c) hybrids by reacting with corresponding halides. For the alternative series of hybrids, the carboxylic acid group of probenecid (10) was similarly elaborated to sulfonamido-1,3,4-oxadiazole-2-thiols (13), and diverged to S-alkylphthalimidated (14a-f) and S-benzylated hybrids (15a-e). Bioactivity assays demonstrated that 8g and 8h are the most potent inhibitors among the synthesized analogs, exhibiting the IC50 values of 13.9 μM and 15.1 μM, respectively. Computational assessment predicted the binding of the inhibitors at an allosteric site developed in the open conformation of DENV2 NS2B/NS3pro. Taken together these findings point out that the synthesized hybrid inhibitors possess a great potential for further antiviral drug development.
中文翻译:

合成和评估新型S-苄基和S-烷基邻苯二甲酰亚胺-恶二唑-苯磺酰胺杂化物作为登革热病毒蛋白酶的抑制剂。
直接作用抗病毒药物(DAAD)成为治疗病毒感染的首选疗法。通过靶向病毒蛋白酶成功开发抗HIV和HCV药物,为发现新型DAAD提供了动力。由两种非结构蛋白NS2B和NS3pro组成的登革热病毒(DENV)蛋白酶也可以用于发现新的抗登革热疗法。在这项研究中,我们将两个药学上有趣的基序(两个替代系列中的1,3,4-恶二唑和苯磺酰胺)连接在一起,以开发新型S-苄基化和S-烷基邻苯二甲酰亚胺化的杂种。对于第一批杂化物,4-氨基苯甲酸(1)通过其氨基与取代的苯磺酰氯反应,而羧酸侧则被精制为磺酰胺基1,3,三步合成4-恶二唑-2-硫醇(6a / b)。在这一阶段,通过与相应的卤化物反应,将中间体6a / b分为S-烷基邻苯二甲酰亚胺化的(8a-j)或S-苄基化的(9a-c)杂化物。对于另一系列的杂种,丙磺舒(10)的羧酸基团类似地被修饰为磺酰胺基-1,3,4-恶二唑-2-硫醇(13),并分歧成S-烷基邻苯二甲酰亚胺基(14a-f)和-苄基化的杂种(15a-e)。生物活性测定表明8g和8h是合成类似物中最有效的抑制剂,IC50值分别为13.9μM和15.1μM。计算评估预测该抑制剂在DENV2 NS2B / NS3pro开放构象中形成的变构位点的结合。
更新日期:2020-01-04
中文翻译:
合成和评估新型S-苄基和S-烷基邻苯二甲酰亚胺-恶二唑-苯磺酰胺杂化物作为登革热病毒蛋白酶的抑制剂。
直接作用抗病毒药物(DAAD)成为治疗病毒感染的首选疗法。通过靶向病毒蛋白酶成功开发抗HIV和HCV药物,为发现新型DAAD提供了动力。由两种非结构蛋白NS2B和NS3pro组成的登革热病毒(DENV)蛋白酶也可以用于发现新的抗登革热疗法。在这项研究中,我们将两个药学上有趣的基序(两个替代系列中的1,3,4-恶二唑和苯磺酰胺)连接在一起,以开发新型S-苄基化和S-烷基邻苯二甲酰亚胺化的杂种。对于第一批杂化物,4-氨基苯甲酸(1)通过其氨基与取代的苯磺酰氯反应,而羧酸侧则被精制为磺酰胺基1,3,三步合成4-恶二唑-2-硫醇(6a / b)。在这一阶段,通过与相应的卤化物反应,将中间体6a / b分为S-烷基邻苯二甲酰亚胺化的(8a-j)或S-苄基化的(9a-c)杂化物。对于另一系列的杂种,丙磺舒(10)的羧酸基团类似地被修饰为磺酰胺基-1,3,4-恶二唑-2-硫醇(13),并分歧成S-烷基邻苯二甲酰亚胺基(14a-f)和-苄基化的杂种(15a-e)。生物活性测定表明8g和8h是合成类似物中最有效的抑制剂,IC50值分别为13.9μM和15.1μM。计算评估预测该抑制剂在DENV2 NS2B / NS3pro开放构象中形成的变构位点的结合。

























 京公网安备 11010802027423号
京公网安备 11010802027423号