当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Elucidating Enzymatic Catalysis Using Fast Quantum Chemical Descriptors.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-10 , DOI: 10.1021/acs.jcim.9b00860 Igor Barden Grillo 1 , Gabriel A Urquiza-Carvalho 2 , José Fernando Ruggiero Bachega 3 , Gerd Bruno Rocha 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-10 , DOI: 10.1021/acs.jcim.9b00860 Igor Barden Grillo 1 , Gabriel A Urquiza-Carvalho 2 , José Fernando Ruggiero Bachega 3 , Gerd Bruno Rocha 1
Affiliation
|
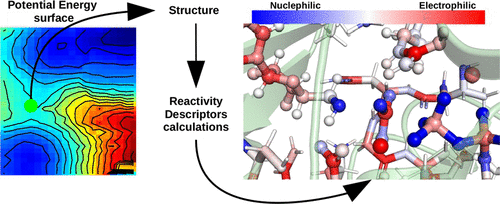
In general, computational simulations of enzymatic catalysis processes are thermodynamic and structural surveys to complement experimental studies, requiring high level computational methods to match accurate energy values. In the present work, we propose the usage of reactivity descriptors, theoretical quantities calculated from the electronic structure, to characterize enzymatic catalysis outlining its reaction profile using low-level computational methods, such as semiempirical Hamiltonians. We simulate three enzymatic reactions paths, one containing two reaction coordinates and without prior computational study performed, and calculate the reactivity descriptors for all obtained structures. We observed that the active site local hardness does not change substantially, even more so for the amino-acid residues that are said to stabilize the reaction structures. This corroborates with the theory that activation energy lowering is caused by the electrostatic environment of the active sites. Also, for the quantities describing the atom electrophilicity and nucleophilicity, we observed abrupt changes along the reaction coordinates, which also shows the enzyme participation as a reactant in the catalyzed reaction. We expect that such electronic structure analysis allows the expedient proposition and/or prediction of new mechanisms, providing chemical characterization of the enzyme active sites, thus hastening the process of transforming the resolved protein three-dimensional structures in catalytic information.
中文翻译:

使用快速量子化学描述符阐明酶催化。
通常,酶催化过程的计算模拟是热力学和结构调查,以补充实验研究,需要高级计算方法来匹配准确的能量值。在目前的工作中,我们提出使用反应性描述子(从电子结构计算出的理论量)来表征酶催化作用,以概述其反应曲线,并使用低水平的计算方法,例如半经验哈密顿量。我们模拟了三种酶促反应路径,其中一种包含两个反应坐标,而无需事先进行计算研究,并计算所有获得的结构的反应性描述符。我们观察到活性部位的局部硬度基本没有变化,对于据说可以稳定反应结构的氨基酸残基,甚至更是如此。这证实了激活能降低是由活性位点的静电环境引起的理论。同样,对于描述原子亲电性和亲核性的量,我们观察到沿反应坐标的突然变化,这也表明酶作为催化反应中的反应物参与。我们期望这样的电子结构分析允许新机制的有利命题和/或预测,提供酶活性位点的化学表征,从而加速催化信息中解析的蛋白质三维结构的转化过程。这证实了激活能降低是由活性位点的静电环境引起的理论。同样,对于描述原子亲电性和亲核性的量,我们观察到沿反应坐标的突然变化,这也表明酶作为催化反应中的反应物参与。我们期望这样的电子结构分析允许新机制的有利命题和/或预测,提供酶活性位点的化学表征,从而加速催化信息中解析的蛋白质三维结构的转化过程。这证实了激活能降低是由活性位点的静电环境引起的理论。同样,对于描述原子亲电性和亲核性的量,我们观察到沿反应坐标的突然变化,这也表明酶作为催化反应中的反应物参与。我们期望这样的电子结构分析允许新机制的有利命题和/或预测,提供酶活性位点的化学表征,从而加速催化信息中解析的蛋白质三维结构的转化过程。这也表明酶作为催化反应中的反应物参与。我们期望这样的电子结构分析允许新机制的有利命题和/或预测,提供酶活性位点的化学表征,从而加速催化信息中解析的蛋白质三维结构的转化过程。这也表明酶作为催化反应中的反应物参与。我们期望这样的电子结构分析允许新机制的有利命题和/或预测,提供酶活性位点的化学表征,从而加速催化信息中解析的蛋白质三维结构的转化过程。
更新日期:2020-01-10
中文翻译:
使用快速量子化学描述符阐明酶催化。
通常,酶催化过程的计算模拟是热力学和结构调查,以补充实验研究,需要高级计算方法来匹配准确的能量值。在目前的工作中,我们提出使用反应性描述子(从电子结构计算出的理论量)来表征酶催化作用,以概述其反应曲线,并使用低水平的计算方法,例如半经验哈密顿量。我们模拟了三种酶促反应路径,其中一种包含两个反应坐标,而无需事先进行计算研究,并计算所有获得的结构的反应性描述符。我们观察到活性部位的局部硬度基本没有变化,对于据说可以稳定反应结构的氨基酸残基,甚至更是如此。这证实了激活能降低是由活性位点的静电环境引起的理论。同样,对于描述原子亲电性和亲核性的量,我们观察到沿反应坐标的突然变化,这也表明酶作为催化反应中的反应物参与。我们期望这样的电子结构分析允许新机制的有利命题和/或预测,提供酶活性位点的化学表征,从而加速催化信息中解析的蛋白质三维结构的转化过程。这证实了激活能降低是由活性位点的静电环境引起的理论。同样,对于描述原子亲电性和亲核性的量,我们观察到沿反应坐标的突然变化,这也表明酶作为催化反应中的反应物参与。我们期望这样的电子结构分析允许新机制的有利命题和/或预测,提供酶活性位点的化学表征,从而加速催化信息中解析的蛋白质三维结构的转化过程。这证实了激活能降低是由活性位点的静电环境引起的理论。同样,对于描述原子亲电性和亲核性的量,我们观察到沿反应坐标的突然变化,这也表明酶作为催化反应中的反应物参与。我们期望这样的电子结构分析允许新机制的有利命题和/或预测,提供酶活性位点的化学表征,从而加速催化信息中解析的蛋白质三维结构的转化过程。这也表明酶作为催化反应中的反应物参与。我们期望这样的电子结构分析允许新机制的有利命题和/或预测,提供酶活性位点的化学表征,从而加速催化信息中解析的蛋白质三维结构的转化过程。这也表明酶作为催化反应中的反应物参与。我们期望这样的电子结构分析允许新机制的有利命题和/或预测,提供酶活性位点的化学表征,从而加速催化信息中解析的蛋白质三维结构的转化过程。


























 京公网安备 11010802027423号
京公网安备 11010802027423号