Current Computer-Aided Drug Design ( IF 1.7 ) Pub Date : 2020-07-31 , DOI: 10.2174/1573409915666190916100437 Amrute B Bhavesh 1 , Amrutkar D Rakesh 2 , Tambe R Santosh 2
|
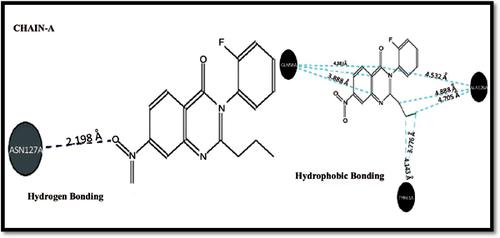
Background: In this present investigation, some 2, 3 disubstituted-quinazolin-4-one derivatives are designed and docked against chain A and chain B of (3WDF) receptor.
Methods: The heterocyclic fused rings quinazolinone have drawn a great attention owing to their expanded applications in the field of pharmaceutical chemistry. The diverse range of molecules with quinazoline/quinazolinone moieties have been reported to exhibit a broad spectrum of biological activities.
Results: The results designate that the quinazolinone ring forms hydrophobic and hydrogen bond contacts with ASN 127 A, ALA 126 A, and SER 83 B, SER 183 B amino acid residue.
Conclusion: Molecular docking is safe and straightforward to use tool which facilitates in investigating, interpreting, enplaning and identification of molecular properties using 3D structures.
中文翻译:
设计和分子对接研究金黄色葡萄球菌UDG的2,3双取代的喹唑啉-4-一个类似物。
背景:在本研究中,设计了约2、3个二取代的喹唑啉-4-酮衍生物,并与(3WDF)受体的链A和链B对接。
方法:杂环稠合环喹唑啉酮由于在药物化学领域的广泛应用而备受关注。据报道,具有喹唑啉/喹唑啉酮基团的分子种类繁多,具有广泛的生物活性。
结果:结果表明,喹唑啉酮环与ASN 127 A,ALA 126 A和SER 83 B,SER 183 B氨基酸残基形成疏水和氢键接触。
结论:分子对接是一种安全,简单易用的工具,它有助于使用3D结构研究,解释,规划和鉴定分子特性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号