


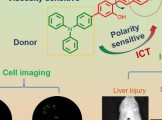
临沂大学杨雷、郑州大学李占先、于明明教授团队CBMI | 利用双信号放大技术诊断肝损伤和评估药物治疗效果的双功能荧光探针
来源:
Chemical & Biomedical Imaging
2024-04-09

































 京公网安备 11010802027423号
京公网安备 11010802027423号