EN
注册
登录
文献直达
发Paper
求职
问答
导师
期刊
资讯
首页
搜索
最新发布
我的收藏
Nano Res.[催化]│青岛科技大学王磊教授团队: 铋纳米颗粒界面改性促进酸性CO₂电还原甲酸
来源: X-MOL
2024-04-18
0
我要评论
取消
发布
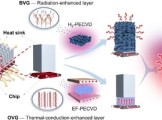
Nano Res.[碳]│北京大学刘忠范团队:通过形貌控制石墨烯功能层实现芯片热管理中传导与辐射的共同增强
来源: X-MOL
2024-04-18
0
我要评论
取消
发布
Nano Res.[催化]│北京化工大学汪乐余教授课题组:超细铂纳米线表面VI B族过渡金属原子位点的配体效应增强氧还原反应性能
来源: X-MOL
2024-04-17
0
我要评论
取消
发布
郑州大学吴翟、苏州纳米所余学超InfoMat:原位制备PtSe2/Ge肖特基结阵列器件实现高灵敏室温宽波段红外探测与成像
来源: X-MOL
2024-04-17
0
我要评论
取消
发布
Nano Res.[能源]│郑州大学凝胶电解质最新进展:构建聚两性离子双网络水凝胶电解质稳定锌金属负极
来源: X-MOL
2024-04-17
0
我要评论
取消
发布
Nano Res.[催化]│北京化工大学汪乐余教授课题组:超细铂纳米线表面VI B族过渡金属原子位点的配体效应增强氧还原反应性能
来源: X-MOL
2024-04-17
0
我要评论
取消
发布
Nano Res.[器件]│打造机器人触觉感官,助力腿足式磁吸附爬壁机器人自主避险
来源: X-MOL
2024-04-16
0
我要评论
取消
发布
Nano Res.[器件]│陈琳/孟佳琳团队:用于神经形态计算的具有超高识别率的有机异质结突触器件
来源: X-MOL
2024-04-16
0
我要评论
取消
发布
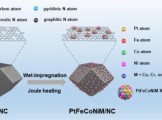
Nano Res.│南方科技大学徐强课题组:MOF辅助合成PtFeCoNiMn纳米高熵合金氧电催化剂增强可充电锌-空气电池性能
来源: X-MOL
2024-04-15
0
我要评论
取消
发布
利用分子自组装策略调控电子诱导的表面反应
来源: X-MOL
2024-04-13
0
我要评论
取消
发布
西安电子科技大学郝跃院士团队常晶晶教授InfoMat:具有高光响应的多功能型p-PCDTBT/n-Ga2O3异质结紫外光电探测器
来源: X-MOL
2024-04-10
0
我要评论
取消
发布
日本东京理科大学Yuichi Negishi教授团队:原子级精确硫醇保护的金纳米团簇——结构设计和物理化学特性的现状总结
来源: X-MOL
2024-04-08
0
我要评论
取消
发布
大连化物所王峰团队/中国海洋大学包锐团队JACS:揭秘海洋飞沫对惰性黑碳的降解和沉降作用
来源: X-MOL
2024-04-03
0
我要评论
取消
发布
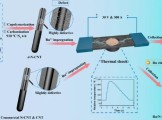
Nano Res.[能源]│王进课题组:热冲击法锚定Ru团簇催化剂,富缺陷助力大电流稳定产氢
来源: X-MOL
2024-03-28
0
我要评论
取消
发布
Nano Res.[半导体]│东南大学徐春祥教授课题组: 表面等离激元增强的自驱动式GaN/ZnTe核壳纳米柱阵列基光电探测器
来源: X-MOL
2024-03-25
0
我要评论
取消
发布
1
2
3
4
5
6
7
8
9
10
11
来看看大家都在关注些什么
有机化学
催化
材料
纳米科技
天然产物
高分子化学
药物与医疗
生命科学
无机化学
物理化学
理论和计算化学
分析化学
环境科学
食品与日用品
工业与商业
职场生涯
轻松生活
热点资讯
RSC主编推荐
盘点
广告
down
wechat
bug
bug




































 京公网安备 11010802027423号
京公网安备 11010802027423号