Glucosamine decreases the stemness of human ALDH+ breast cancer stem cells by inactivating STAT3.
Oncology Letters ( IF 2.5 ) Pub Date : 2018-07-26 , DOI: 10.3892/ol.2018.9222
Rendy Hosea 1 , Novi Silvia Hardiany 1 , Osamu Ohneda 2 , Septelia Inawati Wanandi 1
Cancer stem cells (CSCs) are a subpopulation of cancer cells responsible for tumor maintenance and relapse due to their ability to resist various anticancer effects. Owing to the resistance of CSCs to the effects of targeted therapy, an alternative strategy that targets post-translational glycosylation may be an improved approach to treat cancer as it disrupts multiple coordinated signaling that maintains the stemness of CSCs. Glucosamine acts as an anticancer agent possibly by inhibiting N-linked glycosylation. The aim of the present study was to investigate the effect of glucosamine on the stemness of breast CSCs, which is regulated by signal transducer and activator of transcription 3 (STAT3) signaling. Human aldehyde dehydrogenase-positive (ALDH+) breast CSCs and MCF7 cells were treated with various concentrations (0.25, 1 or 4 mM) of glucosamine for 24 h. Subsequently, cell viability was determined by performing a trypan blue exclusion assay, pluripotency gene [ALDH 1 family member A1 (ALDH1A1), octamer-binding transcription factor 4 (OCT-4), and Krüppel-like factor 4 (KLF4)] expression was determined using the reverse transcription-quantitative polymerase chain reaction, and STAT3 and phosphorylated STAT3 (pSTAT3) levels were determined by performing western blot analysis. Furthermore, the number of mammosphere-forming units (MFUs) in ALDH+ breast CSCs and MCF7 cells was determined. It was determined that glucosamine treatment decreased the viability of ALDH+ breast CSCs. Glucosamine treatment also decreased the stemness of ALDH+ breast CSCs and MCF7 cells, as indicated by decreased ALDH1A1, OCT-4 and KLF4 expression level, and a decreased number of MFUs. This effect of glucosamine may be associated with a decreased pSTAT3/STAT3 ratio, indicating that glucosamine inhibited STAT3 activation; therefore, the results of the present study indicated that glucosamine treatment may be an improved approach to target the stemness of CSCs.
Targeting Transcription Factor YY1 for Cancer Treatment: Current Strategies and Future Directions.
Cancers ( IF 4.5 ) Pub Date : 2023-07-05 , DOI: 10.3390/cancers15133506
Rendy Hosea 1, 2 , Sharon Hillary 1, 2 , Shourong Wu 1, 2, 3 , Vivi Kasim 1, 2, 3
Cancer represents a significant and persistent global health burden, with its impact underscored by its prevalence and devastating consequences. Whereas numerous oncogenes could contribute to cancer development, a group of transcription factors (TFs) are overactive in the majority of tumors. Targeting these TFs may also combat the downstream oncogenes activated by the TFs, making them attractive potential targets for effective antitumor therapeutic strategy. One such TF is yin yang 1 (YY1), which plays crucial roles in the development and progression of various tumors. In preclinical studies, YY1 inhibition has shown efficacy in inhibiting tumor growth, promoting apoptosis, and sensitizing tumor cells to chemotherapy. Recent studies have also revealed the potential of combining YY1 inhibition with immunotherapy for enhanced antitumor effects. However, clinical translation of YY1-targeted therapy still faces challenges in drug specificity and delivery. This review provides an overview of YY1 biology, its role in tumor development and progression, as well as the strategies explored for YY1-targeted therapy, with a focus on their clinical implications, including those using small molecule inhibitors, RNA interference, and gene editing techniques. Finally, we discuss the challenges and current limitations of targeting YY1 and the need for further research in this area.
The two sides of chromosomal instability: drivers and brakes in cancer
Signal Transduction and Targeted Therapy ( IF 40.8 ) Pub Date : 2024-03-29 , DOI: 10.1038/s41392-024-01767-7
Rendy Hosea 1, 2 , Sharon Hillary 1, 2 , Sumera Naqvi 1, 2 , Shourong Wu 1, 2, 3 , Vivi Kasim 1, 2, 3
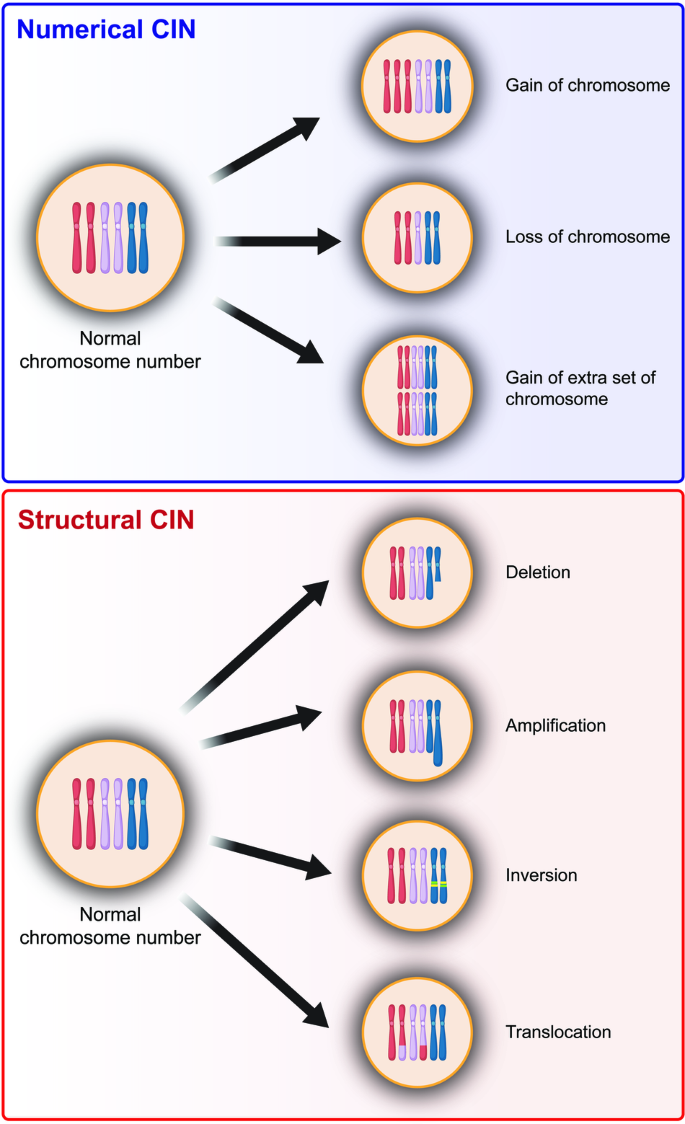 |
Chromosomal instability (CIN) is a hallmark of cancer and is associated with tumor cell malignancy. CIN triggers a chain reaction in cells leading to chromosomal abnormalities, including deviations from the normal chromosome number or structural changes in chromosomes. CIN arises from errors in DNA replication and chromosome segregation during cell division, leading to the formation of cells with abnormal number and/or structure of chromosomes. Errors in DNA replication result from abnormal replication licensing as well as replication stress, such as double-strand breaks and stalled replication forks; meanwhile, errors in chromosome segregation stem from defects in chromosome segregation machinery, including centrosome amplification, erroneous microtubule–kinetochore attachments, spindle assembly checkpoint, or defective sister chromatids cohesion. In normal cells, CIN is deleterious and is associated with DNA damage, proteotoxic stress, metabolic alteration, cell cycle arrest, and senescence. Paradoxically, despite these negative consequences, CIN is one of the hallmarks of cancer found in over 90% of solid tumors and in blood cancers. Furthermore, CIN could endow tumors with enhanced adaptation capabilities due to increased intratumor heterogeneity, thereby facilitating adaptive resistance to therapies; however, excessive CIN could induce tumor cells death, leading to the “just-right” model for CIN in tumors. Elucidating the complex nature of CIN is crucial for understanding the dynamics of tumorigenesis and for developing effective anti-tumor treatments. This review provides an overview of causes and consequences of CIN, as well as the paradox of CIN, a phenomenon that continues to perplex researchers. Finally, this review explores the potential of CIN-based anti-tumor therapy.
YY2/BUB3 Axis promotes SAC Hyperactivation and Inhibits Colorectal Cancer Progression via Regulating Chromosomal Instability
Advanced Science ( IF 14.3 ) Pub Date : 2024-04-29 , DOI: 10.1002/advs.202308690
Rendy Hosea 1, 2 , Wei Duan 1, 2 , Ian Timothy Sembiring Meliala 1, 2 , Wenfang Li 1, 2 , Mankun Wei 1, 2 , Sharon Hillary 1, 2 , Hezhao Zhao 3 , Makoto Miyagishi 4 , Shourong Wu 1, 2, 5 , Vivi Kasim 1, 2, 5
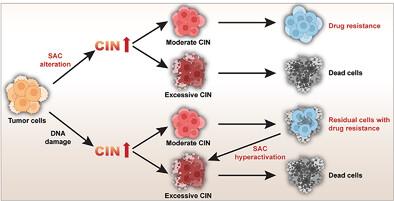 |
Spindle assembly checkpoint (SAC) is a crucial safeguard mechanism of mitosis fidelity that ensures equal division of duplicated chromosomes to the two progeny cells. Impaired SAC can lead to chromosomal instability (CIN), a well-recognized hallmark of cancer that facilitates tumor progression; paradoxically, high CIN levels are associated with better therapeutic response and prognosis. However, the mechanism by which CIN determines tumor cell survival and therapeutic response remains poorly understood. Here, using a cross-omics approach, YY2 is identified as a mitotic regulator that promotes SAC activity by activating the transcription of budding uninhibited by benzimidazole 3 (BUB3), a component of SAC. While both conditions induce CIN, a defect in YY2/SAC activity enhances mitosis and tumor growth. Meanwhile, hyperactivation of SAC mediated by YY2/BUB3 triggers a delay in mitosis and suppresses growth. Furthermore, it is revealed that YY2/BUB3-mediated excessive CIN causes higher cell death rates and drug sensitivity, whereas residual tumor cells that survived DNA damage-based therapy have moderate CIN and increased drug resistance. These results provide insights into the role of SAC activity and CIN levels in influencing tumor cell survival and drug response, as well as suggest a novel anti-tumor therapeutic strategy that combines SAC activity modulators and DNA-damage agents.
Chromosome Missegregation Triggers Tumor Cell Pyroptosis and Enhances Anti‐Tumor Immunotherapy in Colorectal Cancer
Advanced Science ( IF 14.3 ) Pub Date : 2025-02-04 , DOI: 10.1002/advs.202409769
Wei Duan 1, 2 , Rendy Hosea 1, 2 , Lingxian Wang 1, 2 , Cao Ruan 1, 2 , Fuqiang Zhao 1, 2 , Jingyi Liu 1, 2 , Hezhao Zhao 3 , Makoto Miyagishi 4 , Shourong Wu 1, 2, 5 , Vivi Kasim 1, 2, 5
 |
Immune checkpoint inhibitor (ICI) therapy is a promising anti‐tumor therapeutic strategy; however, its efficacy in solid tumors is limited. Chromosome missegregation is common in various solid tumors; however, its role in tumor progression remains poorly understood, and its correlation with ICI is yet to be explored. Here, it is found that increased chromosome missegregation promotes tumor immune microenvironment, and eventually immunotherapeutic efficacy, by triggering pyroptosis. yin yang 2 (YY2) is identified as a mitotic checkpoint regulator, which promotes chromosome missegregation by upregulating BUB1B transcription. Increased chromosome missegregation promoted the formation of micronuclei and release of double‐stranded DNA (dsDNA) into the cytosol, triggering an AIM2‐mediated cytosolic dsDNA response. The subsequent pyroptosis strengthened the tumor immune microenvironment, thereby enhancing immunoinfiltration and cytotoxicity of CD8+ T cells, while preventing their exhaustion. Finally, through in vitro and in vivo experiments, it is demonstrated that combining YY2 overexpression‐induced chromosome missegregation/cytosolic dsDNA response and PD‐1 inhibitor significantly enhanced the efficacy of ICI immunotherapy in microsatellite instable and microsatellite stable colorectal cancer cells. Together, these findings provide new insights on the role of chromosome missegregation in triggering cytosolic dsDNA response‐mediated pyroptosis and modulating the tumor immune microenvironment, suggesting a novel strategy for improving ICI therapeutic efficacy in colorectal cancer.

