
For Gromacs https://webff-documentation.readthedocs.io/en/latest/Reference/NonBond-LJ-GROMACS.html
The non-bond Lennard-Jones GROMACS potential has the functional form:
E=4⋅ϵ⋅[(σRij)12−(σRij)6]+SLJ(Rij)
The force-field parameters for this potential and units are given by:
| Equation Symbol | Parameter Definition | Units |
| ϵ |
Potential well depth for atom [i] | energy/mol |
| σ |
Interatomic cut-off distance for atom [i] | length |

此外,参考http://jerkwin.github.io/GMX/GMXman-5/
原子类型 静态性质 (参见表 5.2)的指定基于几个地方的数据. 质量来源于atomtypes.apt文件(参见5.2.1节), 电荷来源于*.rtp(.rtp = residue topology parameter, 残基拓扑参数, 参见5.6.1节)文件. 这就意味着只对构建氨基酸, 核酸的基本单元定义了电荷, 对其他的分子, 用户需要自己定义电荷. 当使用pdb2gmx程序生成一个拓扑文件(*.top)时, 来自这些文件的信息将被整合在一起.
非键参数包括van der Waals参数V(c6或 σ, 由组合规则决定)和W(c12或 ϵ), 它们列在ffnonbonded.itp文件中, 其中的ptype是粒子类型(参见表 5.1). [ *type ]指令中的条目和键合参数会被应用它们在拓扑文件中的相应部分. 除了将在5.3.4节中提到的那些, 缺少参数将导致警告.
[ atomtypes ]
;name at.num mass charge ptype V(c6) W(c12)
O 8 15.99940 0.000 A 0.22617E-02 0.74158E-06
OM 8 15.99940 0.000 A 0.22617E-02 0.74158E-06
.....
[ nonbond_params ]
; i j func V(c6) W(c12)
O O 1 0.22617E-02 0.74158E-06
O OA 1 0.22617E-02 0.13807E-05
.....注意: GROMACS所包含的大部分力场也带有at.num.列, 但相同的信息位于OPLS_AA bond_type列. 参数V与W的含义取决于拓扑文件[ defaults ]段中选择的组合规则(参见5.7.1节).
对组合规则1:
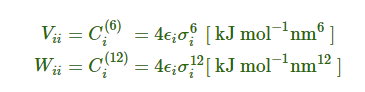
对组合规则2和3:
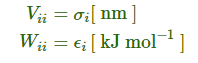
对不同原子类型间的一些或所有组合都可以在[ nonbond_params ]段给出, 参数V与W的定义同上. 对于其他没有给出的任何原子组合, 将根据组合原则, 利用相应原子类型的参数进行计算.
对组合规则1和3:

对组合规则2:

当需要提供 σ 和 ϵ时(规则2和3), 看起来不可能使用非零的 C12 与零值的 C6 参数进行组合. 然而, 提供负值的 σ恰好会这样做, C6 被设为零, C12 正常计算. 这只是代表了读入 σ 值的一种特殊情况, 没有其他的.
对Buckingham势只有一种组合规则:
For Amber
The Amber formulaFon of the 6-12 Lennard-Jones (LJ) potenFal, Vij, between 2 atoms i and j is:


Difference
