
more information see from https://amber-md.github.io/cpptraj/CPPTRAJ.xhtml#subsec_cpptraj_secstruct
rmsd,rmsf,b-factor,radius of gyration,hydrogen bond,database of secondary structure assignments (DSSP),Principal components analysis (PCA),Dynamics cross correlation,Network and community analysis
generate pdb from mdrest5
ambpdb -p com_solvated.top <mdrest5> Full_Mini.pdb
To generate a pdb within 3 A of protein
parm prmtop
trajin mdcrd5.nc lastframe # the last frame (or trajin mdrest5)
autoimage # centering for Periodic Boundary Conditions
mask (:1-300<:3.0) maskpdb mask.pdb # select all residues within 3 Å of residue 1-300
trajout lastframe.pdb pdb #the entire system
run#trajin Test1.crd 10 last 2
#This will process Test1.crd from frame 10 to the last frame, skipping by 2 frames.
note: maestro cannot read the pdb file from cpptraj, try convert with VMD vmd -e input
The input is given here
mol load pdb "Xxx.pdb"
set a [atomselect top "resid 1 to 13955"]
atomselect0 writepdb "convert.pdb"
exit
A script to do this over many files should be constructed...
To produce the readable trajectory file
wrap.in
parm prmtop
trajin mdcrd4.nc
trajout wrap-mdcrd4 trajectory
image origin center familiar com :401
go
cpptraj < wrap.in
##############
rmsd,rmsf,hbond with amber
rmsd.in
parm prmtop
trajin mdcrd5.nc
strip :Na+,Cl-,WAT
autoimage origin
reference crystal.pdb
rms reference mass out protein.rmsd :1-444&!@H=
cpptraj < rmsd.in
xmgrace protein.rmsd
###if you want to run rmsd with a reference structure####
parm prmtop
trajin mdcrd6
strip :Na+,Cl-,WAT
autoimage origin
reference prmcrd
rms reference mass out protein2.rmsd :1-443&!@H=
RMSF
1. strip.in (generate a trajectory with only protein)
parm prmtop
trajin mdcrd5
strip :Na+,Cl-,WAT
#parmstrip :Na+,Cl-,WAT
#parmwrite out new.prmtop
trajout new.mdcrd.nc
2. strip.in (generate a coresponding prmtop)
parm prmtop
#strip :Na+,Cl-,WAT
parmstrip :Na+,Cl-,WAT
parmwrite out new.prmtop
#trajout new.mdcrd
3.rmsf.in
parm new.prmtop
trajin new.mdcrd5.nc
image origin center familiar com :407
rms first
average crdset MyAvg
run
rms ref MyAvg
atomicfluct byres out protein.rmsf :1-444&!@H=
#atomicfluct byres out backbone.rmsf @C,CA,N
cpptraj < rmsf.in
from https://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/render/render.html
Users can easily import structure-related data into Chimera in the form of attributes, or values associated with atoms, residues, or models. The data can be imported with Define Attribute and then represented visually with color ranges, atomic radii, or "worm" thickness. Such data can also be manipulated programmatically in Chimera, and in fact Chimera was designed with extensibility and programmability in mind. It is largely implemented in Python, with certain features coded in C++ for efficiency. Python is an easy-to-learn interpreted language with object-oriented features. All of Chimera's functionality is accessible through Python and users can implement their own algorithms and extensions without any Chimera code changes, so any such custom extensions will continue to work across Chimera releases. Many programming examples are provided to assist extension writers.
https://www.cgl.ucsf.edu/chimera/ImageGallery/framedpecworm.html

The image shows a "worm" representation of pectate lyase C (PDB entry 2pec). The Render by Attribute extension was used to show residue average B-factor values with color (increasing blue to white to red) and worm radius (larger for higher values). Solvent has been deleted. The background and depth-cueing color is sky blue. Subdivision quality was increased to 10 (Effects) and with Shininess, the specular color was changed to white and the brightness was increased to 1.8.
More from https://www.cgl.ucsf.edu/chimera/features.html
hbond
hbond.in
# Hydrogen bond analysis with cpptraj # Load topology and trajectoryparm prmtoptrajin mdcrd5.nc
# All solute-solute and solute-solvent hydrogen bonds + water bridges.
hbond All out All.hbvtime.dat solventdonor :WAT solventacceptor :WAT@O avgout All.UU.avg.dat solvout All.UV.avg.dat bridgeout All.bridge.avg.dat
# Backbone hydrogen bonds only. Save time series data.
hbond Backbone :1-444@C,O,N,H avgout BB.avg.dat series uuseries bbhbond.gnu
# Backbone hydrogen bonds only. Save time series data.
hbond Backbone :1-444@C,O,N,H avgout BB.avg.dat series uuseries bbhbond.gnu
#all hydrogen bonds within residues 1-444, writing the number of Hbonds per frameto "nhb.dat" and
#infromation on each hydrogen bond found to "avghb.dat":
hbond :1-444 out nhb.dat avgout avghb.dat
# to search for all hydrogen bonds formed between donors in residues 1-300 and acceptors in residues 301-500
hbond donormask :1-300 acceptormask :301-500 out nhb.dat avgout avghb.dat
hbond acceptormask :1-300 donormask :301-500 out nhb-.dat avgout avghb-.dat
# Run to actually find hydrogen bonds and generate data. run######################################################################################
#Keep only specified atoms (opposite of strip). This can also be used in conjunction with output from the hbond
#command to retain solute and only bridging residues
For example, the following run
generates bridging data with the ’hbond’ command in a first pass, then uses the bridge ID data to retain only 1
single bridging water between residues 10 and 11:
parm tz2.ortho.parm7
trajin tz2.ortho.nc
# First pass, generate bridge time series
hbond hb solventacceptor :WAT@O solventdonor :WAT out hb.dat
run
# Second pass, retain only frames where the bridge is present
# for residues 10 and 11.
keep bridgedata hb[ID] nbridge 1 bridgeresonly 10,11 parmout keep.parm7
# Write trajectory
trajout keep.nc
run
This run reads in bridge ID data from a previous hbond run and uses it to keep only residues 10, 11, and a bridging water:
parm tz2.ortho.parm7
trajin tz2.ortho.nc
readdata hb.dat
keep keepmask :10,11 bridgedata hb.dat:5 nbridge 1 bridgesonly 10,11 \
parmout keep.10.11.parm7
trajout keep.10.11.nc
run
#######################################################################################
# Perform lifetime analysis on backbone hydrogen bonds
lifetime Backbone[solutehb] out backbone.lifetime.datrunanalysis
cpptraj < hbond.in
Hbond with mdanalysis
https://www.mdanalysis.org/docs/documentation_pages/analysis/hbond_analysis.html
Hbond with PyContact
http://www.ks.uiuc.edu/Research/pycontact/
https://github.com/maxscheurer/pycontact
using VMD
1. open trajectory file 2. Extensions 3. Analysis 4. Hydrogen Bonds 5. Angle Cutoff 20 (maybe 40)6. Write output to file 7. Find hydrogen bonds
distance.in
parm prmtop
trajin mdcrd5
distance end-to-end @1234 @5677 out dist-end-to-end.agr
run
writedata end-to-end.dat end-to-end
cpptraj < distance.in
dssp
parm prmtop
trajin mdcrd5
secstruct :1-444 out dssp.gnu
run
cpptraj < dssp.in
run gnu-form
sed -i '1d' dssp.gnu ###remove the first line
gnuplot dssp.gnu
gnu-form is a script with
sed -i '1,12d' dssp.gnu ###remove the line 1 to 12
cat>>insert.gnu<<EOF
reset
unset key
set grid
set terminal pngcairo
set size 0.96,1
set encoding iso_8859_1
set xtics nomirror
set ytics nomirror
set pm3d map corners2color c1
set cbrange [ -0.500: 7.500]
set cbtics 0.000,1,7
set palette maxcolors 8
set palette defined (0 "white",1 "#0000FF",4 "#00FF00", 5 "grey", 7 "#FF0000")
set cbtics("None" 0.000,"Para" 1.000,"Anti" 2.000,"3-10" 3.000,"Alpha" 4.000,"Pi" 5.000,"Turn" 6.000,"Bend" 7.000)
set xlabel "Time (ns)"
set ylabel "Residue Number"
set yrange [ 0.000: 450] ### if there are 450 residules in the protein
set xrange [ 0.000: 10] ### if the simulation is 10 ns
set title "DSSP of this protein"
set output "dssp.png"
splot "-" u (\$1/1):2:3 w pm3d title "dssp.gnu" ##set 1/1 if your simulation is 10 ns, 1/2 for 20 ns
EOF
sed -i '1 r insert.gnu' dssp.gnu
\rm insert.gnu
sed -i 's/pause -1/set output/g' dssp.gnu
Copy from https://sunxiaoquan.wordpress.com/2016/10/22/dssp-analysis-in-ambertools/
I did the following modification to generate PNG file or PDF file.
The head of GNU file can be changed to the following content.
DSSP: see https://swift.cmbi.umcn.nl/gv/dssp/DSSP_3.html
installation:
sudo apt-get install dssp
。。。。。
reference
1. Structure-Activity Relationship in TLR4 Mutations: Atomistic Molecular Dynamics Simulations and Residue Interaction Network Analysis
2. Amino Acid Oxidation of Candida antarctica Lipase B Studied by Molecular Dynamics Simulations and Site-Directed Mutagenesis
3. Molecular Dynamics Simulations of A27S and K120A Mutated PTP1B Reveals Selective Binding of the Bidentate Inhibitor
Network and community analysis
https://md-task.readthedocs.io/en/latest/index.html
radius of gyration
30.10.58. radgyr | rog
radgyr [name>] [<mask>] [out <filename>] [mass] [nomax] [tensor]
[<name>] Data set name.
[<mask>] Atoms to calculate radius of gyration for; default all atoms.
[out <filename>] Write data to <filename>.
[mass] Mass-weight radius of gyration.
[nomax] Do not calculate maximum radius of gyration.
[tensor] Calculate radius of gyration tensor, output format ’XX YY ZZ XY XZ YZ’.
Data Sets Created:
<name> Radius of gyration in Ang.
<name>[Max] Max radius of gyration in Ang.
<name>[Tensor] Radius of gyration tensor; format ’XX YY ZZ XY XZ YZ’.
Calculate the radius of gyration of specified atoms. For example, to calculate only the mass-weighted radius of
gyration (not the maximum) of the non-hydrogen atoms of residues 4 to 10 and print the results to “RoG.dat”:
gyration.in
parm prmtop
trajin mdcrd5
radgyr :4-10&!(@H=) out RoG.dat mass nomax
run
cpptraj < gyration.in
from paper by Ulf Ryde
Amino Acid Oxidation of Candida antarctica Lipase B Studied by Molecular Dynamics Simulations and Site-Directed Mutagenesis

B-factor与RMSF的关系?
做完分子动力学后,一般用RMSD或RMSF来衡量蛋白质整体骨架的变化。
RMSD 相对与某一构型的均方根偏差,RMSD 降低表明结构更稳定,结合更牢。
RMSF 相对与平均构型的均方根涨落,B-Factors 和RMSF 差不多,他们反映的是结构在
整个模拟中的平均变化情况。RMSF的数值大说明对应的位置的柔性大,反之相反。
RMSF^2=3B/(8*pi^2)
算得RMSF,依此式将RMSF与B-factor 相互转化。
1.制作模拟电影
a)使用VMD
VMD的“extensions->Visualization->Movie Maker"可以直接制作小电影。制作电影时,注意使用VMD的“Trajectory Smoothing", 将模拟轨迹的几个帧(5~10帧)平均。(具体请参考VMD教程)
b)使用pymol
先用g_filter平均轨迹:
g_filter –smd.tpr –f protein.xtc –ol movie.pdb –fit –nf 5
参数:-ol,处理后电影文件,此处为PDB文件格式;-fit,将各帧进行结构平均;-nf,指定平均的相邻帧数。(PS:帧,即frame)。
使用pymol打开电影文件:
pymolmovie.pdb
在pymol里面把分子结构设置成自己喜欢的表示方式,cartoon,surface等等,然后使用一下命令将轨迹的每一帧制作成图片:
mpngframe_.png
该命令会产生一系列以frame开头的,带数字序列号的PNG图片文件。最后使用Linux系统的convert命令,将这些文件连城电影文件即可:
convert -delay15 -loop 0 frame_*.png movie.gif
以上命令行的最后文件,movie.gif,即为小电影文件。其中,-delay参数就是设定每一帧直接的时间间隔。
2.重叠两个结构比较构象变化 with gromacs
g_confrms -f1structure_1.gro-f2 structure_2.gro -o fit.pdb (-n1 index_1.ndx -n2 index_2.ndx)
参数:-f1,-f2,即需要进行比较的两个分子结构;-o,输出合并后分子结构,fit.pdb包含两个分子结构的文件;-n1,-n2,指定索引文件,可以选择那一部分分子进行重叠。
PS: 也可以将两个结构使用editconf转换成PDB文件,两者都加载到pymol中,然后使用align命令将结构直接重叠。
3.计算平均结构 with gromacs
a)g_covar -stopol.tpr -f traj.xtc -b 500 -e 800 -av traj_aveg.pdb
g_covar主要用来计算结构协方差矩阵,也可以用来计算平均结构。以上命令可以得到模拟轨迹中500到800皮秒的分子平均结构,结果输出为traj_aveg.pdb。
b)g_rmsf -stopol.tpr-ftraj.xtc-b500-e800-oxtraj_aveg.pdb
g_rmsf用于计算分子结构均方根涨落,也可以求平均结构,以上命令功能与使用g_covar相当。
4.计算系统能量和相互作用能量 with gromacs
g_energy -stopol.tpr-f traj.xtc -o energy.xvg
该命令为交互方式,需要选择输出的能量,可以仔细阅读各个能量项,输出需要部分(参考软件手册)。输出文件为xmgrace的.xvg文件格式(个人最爱软件之一,理由:简单,强大,免费!!)
5.计算蛋白回转半径 with gromacs
何谓“回转半径”?就是大学物理第二册,第三百页左下角,第二十五行的后半部分。回转半径,可以大概描述分子结构有多紧密。
g_gyrate -stopol.tpr -f traj.xtc -o rgyrate.xvg
可以使用索引文件,指定特定部分分子。
6.计算蛋白质结构变化 with gromacs
a)均方根偏差(Root-Mean-Square-Deviation, RMSD)
g_rms -stopol.tpr -f traj.xtc -o rmsd.xvg
这基本是模拟之后,分析第一步做的事情,主要用于查看模拟结构(特别是蛋白质)是否稳定。RMSD公式如下:
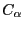
a)均方根涨落(Root-Mean-Square-Fluctuation, RMSD) with gromacs
g_rmsf -stopol.tpr -f traj.xtc -o rmsf.xvg
该命令可以得到每一个原子的结构位置涨落(也就是震动的构象变化)。均方根涨落的公式如下:

其中r_t是某一个时刻的构象,r_ref是参考构象,T则是总时间。RMSF和RMSD都是比较结构变化的工具,两者功能相似,方程相似。RMSD是对原子总数求平均,RMSF是对时间求平均(这个要记住)。
在Gromacs中,g_rms得到的是随时间变化的曲线,g_rmsf得到的是随原子序号变化的曲线(可以使用-res选项得到随氨基酸残基变化的曲线)。相信使用xmgrace打开结构文件一看,地球人基本都明白什么意思。
g_rmsf也可以计算平均结构:
g_rms -s topol.tpr -f traj.xtc -ox traj_ave.pdb -b 800 -e 1000
以上既是求800ps到100ps的估计平均结构,可以与上文使用g_covar求平均结构进行比较。
c)原子位置变化RMSD with gromacs
g_rmsdist -s topol.tpr -f traj.xtc -o distrmsd.xvg
该命令求得的是原子间距离变化的RMSD。也可以使用索引文件选择需要计算的原子。该命令还有很多奇妙输出,请使用g_rmsdist -h查看一下。
(1)选择体系的一部分,以便分析 . 采用make_ndx 命令。
make_ndx -f protein.pdb -o protein.mdx
> r 1 36 37 72 73 108 (按残基选择,还可按其他方式进行选择)
> name 15 Terminal (对选择的这部分残基命令)
> v (查看)
> q (退出)
(2) 分析能量,采用g_energy命令。
g_energy –f md.edr –o potential.xvg
然后选择所有输出的能量部分的序号。
(3) 分析原子的均方根偏差(RMSD),采用g_rms命令。
g_rms –s md.tpr –f md.trr –o rmsd.xvg
(4) 分析蛋白质的回旋半径变化,采用g_gyrate命令。
蛋白质的回旋半径反应了蛋白质分子的体积和形状。同一体系的回旋半径越大,说明体系发生了膨胀。
g_gyrate –f md.trr –s md.tpr –o gyrate.xvg
(5) 分析溶剂的可及表面积(SASA),采用g_sas命令。
它是描述蛋白质疏水性的重要手段,氨基酸残基的疏水性是影响蛋白质折叠的重要物理作用。
g_sas -f md.trr -s md.tpr -o area.xvg -or resarea.xvg -oa atomarea.xvg
(6) 分析蛋白质的二级结构变化,采用do_dssp命令。
do_dssp –s md.tpr –f md.xtc –o ss.xpm