
Abstract
Telomere shortening is associated with idiopathic pulmonary fibrosis (IPF), a high-morbidity and high-mortality lung disease of unknown etiology. However, the underlying mechanisms remain largely unclear. In this study, wild-type (WT) mice with normal telomeres and generation 3 (G3) or G2 telomerase RNA component (TERC) knockout Terc−/− mice with short telomeres were treated with and without lipopolysaccharide (LPS) or bleomycin by intratracheal injection. We show that under LPS induction, G3 Terc−/− mice develop aggravated pulmonary fibrosis as indicated by significantly increased α-SMA, collagen I and hydroxyproline content. Interestingly, TGF-β/Smads signaling is markedly activated in the lungs of G3 Terc−/− mice, as indicated by markedly elevated levels of phosphorylated Smad3 and TGF-β1, compared with those of WT mice. This TGF-β/Smads signaling activation is significantly increased in the lungs of LPS-treated G3 Terc−/− mice compared with those of LPS-treated WT or untreated G3 Terc−/− mice. A similar pattern of TGF-β/Smads signaling activation and the enhancing role of telomere shortening in pulmonary fibrosis are also confirmed in bleomycin-induced model. Moreover, LPS challenge produced more present cellular senescence, apoptosis and infiltration of innate immune cells, including macrophages and neutrophils in the lungs of G3 Terc−/− mice, compared with WT mice. To our knowledge, this is the first time to report telomere shortening activated TGF-β/Smads signaling in lungs. Our data suggest that telomere shortening cooperated with environment-induced lung injury accelerates the development of pulmonary fibrosis, and telomere shortening confers an inherent enhancing factor to the genesis of IPF through activation of TGF-β/Smads signaling.
Similar content being viewed by others


Introduction
Idiopathic pulmonary fibrosis (IPF) is a devastating progressive lung disease of unknown origin. It is characterized by the deposition of excessive collagen in interstitial spaces between alveoli, which aggressively destroys normal lung architecture and results in severe lung function decline [1]. This disease has a median survival of 2–5 years after diagnosis, and no effective therapies are available to date [2]. IPF usually affects older adults; the disease's incidence is age dependent [3]. Although little is known about its etiology, genetic studies have provided clues to its molecular mechanisms [4,5,6]. It has been found that in 19% of patients with familial pulmonary fibrosis (FPF) and 3% of patients with sporadic pulmonary fibrosis (SPF), the pathological gene arose from mutations to telomerase reverse transcriptase (TERT) or telomerase RNA component (TERC), resulting in insufficient or deficient telomerase activities and significantly shortened telomeres [4, 7,8,9,10]. However, the mutation rate does not adequately explain why, in 40% of patients with FPF and 20% of patients with SPF, telomere lengths were significantly shortened. This suggests that mutations occurred in regulation regions of telomerase other than coding regions, or that other gene mutations can also affect telomere length [7, 9, 11]. Until now, the underlying mechanism of how telomere shortening causes IPF has been largely unknown.
TERT and TERC are two essential factors of telomerase, a remarkable enzyme that mainly catalyzes the addition of repetitive DNA to the shortening telomere due to the end replication problem in each cell division [12,13,14]. High telomerase activity is usually found in somatic stem cells, germ cells and immortalized cancer cells [15, 16]. In contrast to TERT, which has multiple functions, TERC's sole function may be to control telomere length; thus, TERC knockout mice may supply an effective model in which to learn how telomere shortening leads to IPF [17,18,19]. Because mice have longer telomeres than humans, later generations of Terc−/− mice are characterized in both theory and practice by short telomeres [19, 20]. Previous studies on these generations did not find significant automatic pulmonary fibrosis, but alveolar integrity and lung regeneration capacity were affected [21, 22]. This suggests the development of pulmonary fibrosis may require other initial factors such as lung injury created by harmful environmental exposures [21, 22]. However, bleomycin, an agent frequently used to induce pulmonary fibrosis, did not produce exacerbated pulmonary fibrosis in late generations of Terc−/− mice compared with wild-type (WT) mice, as was expected [23].
Lipopolysaccharide (LPS), derived from the outer membrane of gram-negative bacteria, has been widely used to establish a model of acute lung injury in which inflammatory response was stimulated [24,25,26,27,28]. Moreover, a number of researchers have used LPS to successfully induce lung fibrosis in both rats and mice [24, 29, 30]. In this study, we applied LPS or low-dose bleomycin in late genereation Terc−/− mice to study the impact of telomere shortening in lung fibrosis.
Materials and Methods
Mice
We interbred Terc+/- mice [31] (a gift from Dr. Zhen-yu Ju) of C57BL/6J background to generate first-generation (G1) Terc−/− mice. We then further interbred these G1 Terc−/− mice to produce second-generation (G2) and third-generation (G3) Terc−/− mice. We used C57BL/6J mice as WT controls. Male and female mice (8–12 weeks) weighing 20–25 g were used for the experiments. They were housed in the experimental animal center of Hangzhou Normal University, Xiasha Campus, where food and water were granted freely. The university's Animal Ethics Committee approved all animal experimental protocols.
LPS or bleomycin administration
We prepared the LPS working solution by dissolving LPS powder, L2880 (Sigma-Aldrich, St. Louis, Missouri, USA) in sterile phosphate-buffered saline (PBS). We administered LPS to G3 Terc−/− and WT mice by intratracheal injection at 5 mg/kg body weight and in a volume of 2 μL/g body weight. We prepared the diluted bleomycin working solution in PBS from bleomycin stocking solution, which is prepared by dissolving bleomycin powder, B5507 (Sigma-Aldrich, St. Louis, Missouri, USA) in 0.1 M potassium phosphate. We administered bleomycin to G2 Terc−/− and Terc+/+ mice by intratracheal injection at 0.3 mg per kg body weight and in a volume of 1.8 μL per gram body weight. Control mice received intratracheal injections of sterile PBS in a volume of 2 μL/g body weight for LPS model, or did not receive any solution for bleomycin model. Mouse lung tissues were excised at 7 days post-LPS injection or 14 days post-bleomycin injection for further analyses.
Histological staining and semi-quantitative score analysis
We fixed harvested lung tissues in Bouin's fixative for 6–8 h at room temperature, then embedded them in paraffin and cut them into sections of 5 μm thick. Then we stained these sections with Masson's trichrome, using HT-15 Trichrome Stain Kit (Sigma-Aldrich, St. Louis, Missouri, USA) according to the manufacturer's instructions, and with hematoxylin-eosin (H&E) (ZSGB-Bio, Beijing, China). We then scanned the trichrome-stained histological specimens at ×20 magnification and randomly acquired 10 sequential non-overlapping fields under ×200 magnification to perform quantification analysis of fibrosis scores. We evaluated the extent of lung fibrosis as previously described [32].
Immunohistochemistry (IHC)
We regularly dewaxed, rehydrated, and treated the paraffin-embedded lung sections with 3% H2O2 in methanol to block endogenous peroxidase. To retrieve antigens, we microwaved the sections in 10 mM citrate buffer, pH 6.0, for 30 min. After cooling them to room temperature, we added an avidin/blocking solution prepared from Avidin/Biotin Blocking Kit SP-2001 (Vector Laboratories, Burlingame, California, USA) to the sections for 50 min, then incubated them with the following primary antibodies in the aforementioned biotin/blocking solution for 60 min at room temperature:
-
Mouse anti-human α-smooth muscle actin (α-SMA), M0851 (1:100; Agilent Pathology Solutions [formerly Dako], Carpinteria, California, USA)
-
Rabbit anti-mouse Anti-Collagen I, ab21286 (1:400; Abcam, Cambridge, UK)
-
Mouse anti-mouse proliferating cell nuclear antigen (PCNA), 2586S (1:2000; Cell Signaling Technology, Danvers, Massachusetts, USA)
-
Rabbit anti-mouse p21, ab2961 (1:50; Abcam, Cambridge, UK)
-
Rat anti-mouse anti-neutrophil, ab2557 (1:200; Abcam, Cambridge, UK)
-
Rat anti-mouse F4/80, MCA497R (1:200; Bio-Rad Antibodies [formerly AbD Serotec], Oxford, UK)
-
Rabbit anti-mouse NF-κB p65 phospho S536, ab86299 (1:50; Abcam, Cambridge, UK)
After rinsing them with PBS 3 times, we incubated the sections for 60 min at room temperature with the following biotinylated secondary antibodies (both from Vector Laboratories, Burlingame, California, USA) in blocking solution: horse anti-rabbit, BA-1100 (1:200) and goat anti-rat, BA-9400 (1:200). We then applied the VECTASTAIN ABC HRP standard kit, PK-4000 (Vector Laboratories, Burlingame, California, USA) to expand positive signaling. Results were visualized using the DAB peroxidase (HRP) substrate kit (with nickel), 3,3′-diaminobenzidine, SK-4100 (Vector Laboratories, Burlingame, California, USA), followed by nucleic counterstain with hematoxylin if necessary. Sections were scanned with a Pannoramic MIDI II system (3DHISTECH, Budapest, Hungary). We counted or quantified the stained cells or stained area percentage with NIH ImageJ software, v.1.48u4. Number of stained cells per field or stained area percentage under ×20 or ×40 magnification was expressed as a mean value ± SE for each mouse lung, based on 10 random images analyzed.
Measurement of lung hydroxyproline content
We determined lung hydroxyproline content with Hydroxyproline Assay Kit, A030-2 (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's instructions.
Western immunoblotting
We prepared proteins from mouse lung tissue in RIPA lysis buffer, subjected samples to standard SDS-PAGE under reducing conditions with antibodies, including Beta-Actin (EM21002, HuaAn Biotechnology, Hangzhou, China), Phospho-Smad3 (Ser204) (PA5-36877, Thermo Fisher Scientific [formerly Invitrogen], Waltham, Massachusetts, USA), α-SMA (M0851, Agilent Pathology Solutions, [formerly Dako]), PCNA (2586S, Cell Signaling Technology), p21 (sc-471-G, Santa Cruz Biotechnology, Dallas, Texas, USA), TGF-β1 (sc-146, Santa Cruz Biotechnology), Neutrophil (ab2557, Abcam), anti-NF-κB p65 (phospho S536) (ab86299, Abcam).
Double immunofluorescence staining
We processed the lung cryosections using standard procedures and permeabilized them with 0.5% sodium dodecyl sulfate (SDS) for 15 min at room temperature. After blocking them with 5% normal donkey serum for 1 h, we incubated the samples overnight at 4 °C, each with one of the following antibodies:
-
Goat anti-mouse SPC, sc-7706 (1:50; Santa Cruz Biotechnology, Inc., Dallas, Texas, USA)
-
Rabbit anti-Cleaved Caspase-3 (Asp175), #9661 (1:200; Cell Signaling Technology, Danvers, Massachusetts, USA)
After washing them fully in phosphate-buffered saline with Tween-20 (PBST), we incubated the sections with Alexa Fluor 488-conjugated donkey anti-goat IgG, A-11055 and Alexa Fluor 555-conjugated donkey anti-rabbit IgG, A-31572 (both 1:200; both from Molecular Probes, Eugene, Oregon, USA) for 1 h at room temperature. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI), and slides were sealed with the mounting medium to prevent fluorescence from weakening. We obtained immunofluorescence images with a Carl Zeiss upright fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany). We counted the positive cells using NIH ImageJ software, v.1.48u4. Number of stained cells per field under ×20 magnification was expressed as a mean value ± SEM for each mouse lung, based on 4 random images analyzed.
Senescence-associated β-galactosidase (SA-β-gal) staining
We performed SA-β-gal staining of the lung tissue with Senescence Cells Histochemical Staining Kit, CS0030 (Sigma-Aldrich, St. Louis, Missouri, USA) according to the manufacturer's instructions. Briefly, we fixed the lung cryosections in ×1 fixation buffer for 6 min at room temperature. After rinsing them with PBS for three times, we incubated the samples with staining mixture at 37 °C without CO2 overnight or longer until the senescence cells were stained blue. On the second day, we adequately rinsed the sections with PBS to remove uncombined crystallization. Slides were counterstained with Nuclear Fast Red, H-3403 (Vector Laboratories, Burlingame, California, USA) for 30 min at room temperature, rinsed well under running water, dried and mounted. We observed the stained slides under a Carl Zeiss regular microscope (Carl Zeiss AG, Oberkochen, Germany). Sections were scanned and images captured with a Pannoramic MIDI II system (3DHISTECH, Budapest, Hungary). We counted the cells expressing β-galactosidase with NIH ImageJ software, v.1.48u4. Number of stained cells per field under ×20 magnification was expressed as a mean value ± SE for each mouse lung, based on 10 random images analyzed.
Sepsis-induced acute lung injury
The procedure of sepsis-induced acute lung injury was followed in the protocol to mild cecal ligation and puncture (CLP) in mice, as described elsewhere previously [33]. Sixteen hours later, we collected mouse lungs to perform real-time quantitative PCR analysis.
Statistical analysis
Results are presented as mean ± SE and are based on the indicated number of mice per group. We assessed statistical significance of differences by two-tailed Student's t test or one-way analysis of variance (ANOVA) using GraphPad Prism v6.0. We considered differences between groups to be significant when P < 0.05, and statistical significance is indicated by P < 0.05, P < 0.01, and P < 0.001.
Results
Both LPS and bleomycin-induced pulmonary fibrosis is significantly increased in telomere shortening mice compared with WT mice
To investigate the effect of telomere shortening on LPS-induced lung fibrosis, we administered 1 dose of LPS, 5 mg/kg, to G3 Terc−/−, and WT mice by intratracheal instillation. After 7 days, we performed histology using H&E and Masson's trichrome staining. LPS-treated G3 Terc−/− mouse lungs showed more-severe pulmonary interstitial edema, inflammatory cell infiltration, alveolar wall thickening and blue collagen fiber deposition than did lungs from any of the other 3 experimental groups (Fig. 1a), indicating the severity of lung injury and tissue fibrosis in LPS-treated G3 Terc−/− mouse lungs.

LPS-induced pulmonary fibrosis was increased in G3 Terc−/− mice compared with WT mice. Seven days after intratracheal instillation of LPS or controlled PBS, we performed H&E staining, Masson's trichrome staining and α-SMA, and collagen I IHC on lung sections from all 4 mouse groups. a Representative images of lung fibrosis score and IHC of lung fibrosis marker α-SMA and collagen I. Upper row indicates H&E staining. Second row indicates Masson's trichrome staining; blue indicates collagen fiber deposition. Third row indicates α-SMA expression in lungs; brown indicates the α-SMA-positive area. Last row indicates collagen I expression in lungs; brown indicates the collagen I-positive area. Square in upper-right corner shows local enlargement of background images. Scale bar = 0.1 mm. b Semiquantitative analysis of lung fibrosis histological score, (c) percentage of α-SMA-positive area, (d) α-SMA-positive myofibroblast count per field, and (e) percentage of collagen I-positive area in all 4 mouse groups. Results are presented as mean ± SE values based on 4–5 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Next, we investigated the degree of lung fibrosis in all 4 groups. We found that fibrosis marker α-SMA was stained in pulmonary vascular smooth-muscle cells (SMCs), bronchial SMCs and myofibroblasts. In the lungs, myofibroblasts are located in the interstitial spaces of alveoli and are regarded as a source of excessive matrix deposition (Fig. 1a). As expected, LPS induced significant pulmonary fibrosis in WT mouse lungs, as indicated by increased semi-quantitative fibrosis score, quantitative α-SMA and collagen I in IHC-stained areas, and number of myofibroblasts (Fig. 1a–e). Interestingly and importantly, as demonstrated by the same indicators, LPS-treated G3 Terc−/− mouse lungs showed a significant increase in pulmonary fibrosis compared with LPS-treated WT mouse lungs (Fig. 1a–e), indicating that telomere shortening aggravates LPS-induced pulmonary fibrosis. Moreover, untreated G3 Terc−/− mouse lungs showed significant increases in semi-quantitative fibrosis scores, α-SMA and collagen I in IHC-stained areas, and myofibroblasts compared with untreated WT mouse lungs. However, they did not show significant alveolar wall thickening, suggesting that the G3 Terc−/− lungs delveloped limited pulmonary fibrosis automatically.
The significantly increased lung fibrosis marker α-SMA in LPS-treated G3 Terc−/− lungs were confirmed by Western immunoblotting analysis (Fig. 2). Moreover, we measured the lung hydroxyproline content, another fibrosis marker in four groups. As expected, LPS-treated G3 Terc−/− lungs showed a significantly increased amount compared with LPS-treated WT lungs (Fig. 3), this is agreeing with the above analyses indicating telomere shortening aggravated LPS-induced lung fibrosis.

Western blot analyses of fibrosis marker α-SMA, TGF-β/Smads signaling proteins phosphorylated Smad3 and TGF-β1 in four groups of LPS model

Measurement of lung hydroxyproline content, a collagen-related index in four groups of LPS model. Results are presented as mean ± SE values based on 3–4 mice per group. ****P < 0.0001
In order to confirm the enhanced role of telomere shortening in lung fibrosis, we tested the impact of telomere shortening in bleomycin-induced lung fibrosis, a broadly used traditional lung fibrosis model. As expected, we observed that α-SMA and collagen 1a1, 3a1 levels were significantly increased in bleomycin-treated G2 Terc−/− lungs compared with bleomycin-treated Terc+/+ lungs (Fig. 4, Supplemental Fig. 1), indicating that telomere shortening also aggravated bleomycin-induced lung fibrosis.

Western blot analyses of fibrosis marker α-SMA, TGF-β/Smads signaling proteins phosphorylated Smad3, cell proliferation index PCNA, cellular senescence index p21, neutrophil marker Ly-6C/6G, and NF-κB signaling protein phosphorylated NF-κB p65 in lungs of 4 mouse groups in bleomycin (BLM) model
Telomere shortening activated TGF-β/Smads signaling, whose activation is significantly increased in LPS or bleomycin-treated telomere shortening mouse lungs
In order to understand the underlying mechanism of telomere shortening enhancing LPS- or bleomycin-induced lung fibrosis, we investigated the activation of TGF-β/Smads signal in four groups of LPS model and four groups of bleomycin model. Surprisingly, in LPS model, untreated G3 Terc−/− lungs showed a markedly elevated phosphorylated Smad3 levels compared with untreated WT lungs, the latter only revealed a very weak or undectable levels (Fig. 2). This demonstrated that the telomere shortening activated the TGF-β/Smads signaling. In addition, LPS-treated G3 Terc−/− lungs showed a significantly increased phosphorylated Smad3 levels whenever compared with untreated G3 Terc−/− lungs or LPS-treated WT lungs (Fig. 2), which indicated that TGF-β/Smads signaling activation was significantly increased in LPS-treated G3 Terc−/− lungs. TGF-β1, a ligand of TGF-β/Smads signaling pathway, whose levels were weakly expressed in untreated WT lungs, while they were strongly expressed in untreated G3 Terc−/− lungs and other two LPS-treated groups (Fig. 2). Moreover, a similar pattern of TGF-β/Smads signaling activation was also observed in the four groups of bleomycin model (Fig. 4).
Telomere shortening aggravates LPS-induced cellular senescence in the lungs
Recent studies have revealed that LPS can also induce cellular senescence in vitro [34]. Cell senescence is a type of cell cycle arrest with functional retrogression. Senescent cells usually express a high level of cyclin-dependent kinase inhibitors, such as p21 and p16 in nuclei, and SA-β-gal in lysosomes in cytoplasm [35]. We evaluated whether telomere shortening or LPS could induce cell senescence in our mouse lungs. Interestingly, we observed that both SA-β-gal-positive stained cells and p21-positive stained cells were significantly increased in LPS-treated WT mouse lungs compared with untreated WT mouse lungs, whereas PCNA-positive stained cells were significantly decreased in LPS-treated WT mouse lungs compared with untreated WT mouse lungs (Fig. 5a–d). Senescent cells were therefore significantly richer in LPS-treated WT than in untreated WT mouse lungs, indicating that LPS induces significant cellular senescence in normal lung tissues. Similarly, LPS induced significant cellular senescence in G3 Terc−/− mouse lungs (Fig. 5a–d). However, the cellular senescence is significantly increased in LPS-treated G3 Terc−/− mouse lungs than LPS-treated WT mouse lungs (Fig. 5a–f), indicating that telomere shortening aggravates cellular senescence induced by LPS in lungs. As expected, we also noticed a significantly increased cellular senescence in untreated G3 Terc−/− mouse lungs than in untreated WT mouse lungs (Fig. 5a–f), suggesting that telomere shortening triggers significant cellular senescence in lung tissues. Moreover, similar results were also observed in bleomycin-treated model as indicated by PCNA and p21 levels (Fig. 4).

Telomere shortening aggravated LPS-induced cellular senescence in the lungs. a Representative images from IHC of PCNA, p21 and SA-β-gal staining in lung sections from 4 mouse groups. Upper row indicates immunostaining of proliferation marker PCNA. Second row indicates SA-β-gal staining (blue); counterstained nuclei (red) are shown by Fast Red staining. Lower row indicates immunostaining of cell cycle inhibitor p21. Square in upper-right corner shows local enlargement of background images. Scale bar = 0.05 mm. Counts of (b) percentage of PCNA stained cells, (c) SA-β-gal stained cells and (d) p21 stained cells per field in lung sections from all 4 mouse groups. e PCNA levels and (f) p21 levels in lungs from all 4 mouse groups by Western blot analysis. Results are presented as mean ± SE values based on 4–5 mice per group. *P < 0.05, ***P < 0.001, ****P < 0.0001
Under LPS treatment, apoptosis is more-present in telomere shortening mice compared with WT mice
To further determine whether telomere shortening would confer genetic susceptibility to LPS treatment, we investigated cell apoptosis in G3 Terc−/− and WT mouse lungs under LPS treatment as measured by cleaved-Caspase-3 immunofluorescence. LPS-treated G3 Terc−/− mouse lungs showed a significant increased apoptotic cells than did LPS-treated WT mouse lungs (Fig. 6a, b), suggesting telomere shortening confers genetic susceptibility to LPS challenge in lung injury. Untreated G3 Terc−/− mice also showed a significant increased apoptotic lung cells than did WT mice (Fig. 6a, b), suggesting that telomere shortening triggers cell apoptosis in the lungs. However, the LPS-treated WT mice showed a slightly increased apoptotic lung cells than did WT mice, suggesting only LPS treatment has limited impact on the lung cell apoptosis compared with LPS combined with the genetic deficiency.

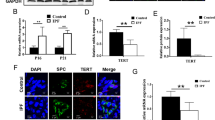
AT2 apoptotic rate was significantly increased in LPS-treated G3 Terc−/− mice than in LPS-treated WT mice. a Representative cleaved-Caspase-3 (red) and SPC (green) double-immunofluorescence images in lung sections from all 4 mouse groups. Nuclei are shown by DAPI (blue) staining; AT2 cells are shown by SPC (green) staining. Scale bar = 0.1 mm. b Count of cells that were positive for cleaved Caspase-3 per field in lung sections from all 4 groups. c Rate of cells that were cleaved-Caspase-3/SPC-double-positive in SPC-positive cells in lung sections from all 4 groups. d Rate of cells that were cleaved-Caspase-3-positive/SPC-negative in lung sections from all 4 groups. The data are represented as mean ± SE values based on 4–5 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Alveolar type II (AT2) cells are stem cells in the alveolar epithelium that help replenish injured epithelial cells [36]. Recent studies have, in fact, suggested that AT2 cells were multipotent adult stem cells in lung tissue [37, 38]. In light of this, we used AT2-specific SPC /cleaved-Caspase-3 double immunofluorescence to monitor AT2 apoptotic rates in mouse lungs from all 4 groups: untreated WT, untreated G3 Terc−/−, LPS-treated WT, and LPS-treated G3 Terc−/−. LPS-treated G3 Terc−/− mouse lungs showed a significant increased AT2 apoptotic rate than did LPS-treated WT mouse lungs (Fig. 6a, c). We also noticed that LPS induced a greater increase in AT2 apoptotic rate in G3 Terc−/− lungs than in WT lungs (Fig. 6c). These results suggest that telomere shortening confers genetic susceptibility to LPS challenge in AT2 cell apoptosis. Moreover, in G3 Terc−/− lungs compared with WT lungs after LPS treatment, cells other than AT2 did not showed a great increase in apoptotic rates (Fig. 6a, d), which may need more investigation.
Telomere shortening exacerbates LPS-induced inflammatory cell infiltration into the lungs
Previous studies have demonstrated that LPS exposure can lead to lung inflammation [39,40,41]. The responsible mechanism is the activation of alveolar macrophages, which release chemokines and cytokines, which in turn recruit neutrophils from peripheral blood to injured lung tissues [39, 42,43,44,45]. To evaluate whether telomere shortening could further contribute to innate pulmonary immune reaction induced by LPS, we examined macrophages and neutrophils in our mouse lungs by detecting their specific markers, F4/80 and Ly-6C/6G, respectively, via immunostaining and Western blot. As expected, LPS induced significant increases in both macrophages and neutrophils in WT mouse lungs (Fig. 7a–d). Interestingly, LPS induced similarly significant increases in such cells in G3 Terc−/− mouse lungs (Fig. 7a–d), and, even without treatment, G3 Terc−/− mouse lungs showed significant increases in both macrophages and neutrophils compared with WT mouse lungs. This indicates that telomere shortening can induce lung inflammation. It has been reported that the number of CD11b+ and CD34+ immune cells was increased in telomerase-deficient lungs, which may be caused by cell senescence induced by telomere shortening [46]. Moreover, LPS-treated G3 Terc−/− mouse lungs showed significantly more macrophages or neutrophils compared with our other 3 groups (Fig. 7a–d), indicating that the effect of LPS superimposed on telomere shortening. Taken together, our results suggest that telomere shortening plays an important role in promoting innate immune response and in recruiting inflammatory cells in lung tissue exposed to LPS. Moreover, similar results were also observed in bleomycin-treated model as indicated by neutrophil levels (Fig. 4).

Telomere shortening exacerbated LPS-induced inflammatory cell infiltration into the lungs. a Representative images of IHC staining from macrophage marker F4/80 and neutrophil marker Ly-6C/6G in lung sections from all 4 mouse groups; upper row points to F4/80-positive staining, lower row to Ly-6C/6G-positive staining. Square in upper-right corner shows local enlargement of background images. Scale bar = 0.05 mm. Counts of (b) macrophage F4/80 cells and (c) neutrophil Ly-6G/6C cells per field in lung sections from all 4 mouse groups. d Western blot analysis of neutrophil Ly-6G/6C levels in lungs from all 4 mouse groups. Results are presented as mean ± SE values based on 4–5 mice per group. *P < 0.05, **P < 0.01, ****P < 0.0001
NF-κB activation is significantly increased in LPS-treated telomere shortening mice
Activation of NF-κB signaling is associated with nuclear translocation of the p65 subunit [47, 48]. In this study, to investigate whether telomere shortening enhances LPS-mediated NF-κB activation, we subjected the frozen mouse lung sections to IHC to indicate phosphorylated NF-κB p65 protein levels. We observed phosphorylated NF-κB p65 in the nuclei of alveolar cells; stain intensity was the weakest in untreated WT, moderate in untreated G3 Terc−/− or LPS-treated WT, and strongest in LPS-treated G3 Terc−/− mouse lungs (Fig. 8a). Similarly, quantitative analysis showed that cells stained positive for nuclear translocation of the p65 subunit were most numerous in LPS-treated G3 Terc−/−, in moderate numbers in untreated G3 Terc−/− or LPS-treated WT and fewest in untreated WT mouse lungs (Fig. 8a, b). These results indicated that NF-κB activation was significantly increased in LPS-treated G3 Terc−/− mouse lungs. Such lungs also showed significantly more cells stained positive for phosphorylated p65 than did LPS-treated WT mouse lungs (Fig. 8a, b), suggesting that telomere shortening further activates LPS-induced NF-κB activation. Moreover, similar results were also observed in bleomycin-treated model, as indicated by phosphorylated p65 levels (Fig. 4).

NF-κB activation was significantly increased in LPS-treated G3 Terc−/− mouse lungs. a IHC staining of phosphorylated NF-κB p65 in lung sections from all 4 mouse groups. Brown indicates cell nuclei that were positive for phosphorylated NF-κB p65. Square in upper-right corner shows local enlargement of background images. Scale bar = 0.05 mm. b Count of cells that were positive for phosphorylated NF-κB p65 per field in lung sections from all 4 groups. c Western blot analysis of phosphorylated NF-κB p65 levels in lungs from all 4 groups of LPS model. Results are presented as mean ± SE values based on 4–5 mice per group. ****P < 0.0001
Telomere shortening in sepsis-induced acute lung injury
To understand the role of telomere shortening in lung injury other than LPS-induction, we set up sepsis-induced acute lung injury by mild CLP procedure. We observed the cytokine IL-6 level was increased in the lungs of G2 Terc−/− mice compared with those of WT mice. CLP-treated G2 Terc−/− mouse lungs showed an increased IL-6 level compared with those of CLP-treated WT mice, suggesting that telomere shortening can enhance sepsis-induced acute lung injury (Supplemental Fig. 3).
Discussion
Although it has been long known that the pathology of a subset of IPF cases is due to telomerase mutations or deliberately induced telomere shortening, the underlying mechanisms of how telomere dysfunction leads to pulmonary fibrosis remain largely unknown. Solving this puzzle would be helpful in developing effective therapeutic strategies for this deadly disease. Meanwhile, in the studies of FPF, it has been demonstrated that some familial members with short telomeres did not suffer from pulmonary fibrosis, suggesting that environment factor play a role in the genesis of IPF [7, 11] We hypothesize that the genetic deficiency with short telomeres combined with environmental factor would lead to the onset of IPF, and telomere shortening would accelerate pulmonary fibrosis mediated by some unknown tissue niches. In our study, we have applied both LPS and bleomycin to induce pulmonary fibrosis. LPS has been found to successfully induce lung fibrosis in animal models; administering it intratracheally at 10 mg/kg or 20 mg/kg body weight in rats [29, 30] and at 3 mg/kg or 5 mg/kg body weight in mice [24, 27] has induced lung fibrosis. In our own study, we have consistently shown that 5 mg/kg LPS administered intratracheally induces lung fibrosis in both WT and telomere shortening mice, as indicated by significantly increased lung fibrosis scoring, Masson's trichrome stain, myofibroblast differentiation, and depositions of collagen I from pathological analysis, as well as by significantly increased lung hydroxyproline content and α-SMA from biochemical analysis. The significantly higher degree of lung fibrosis in LPS-treated telomere shortening mice than in LPS-treated WT mice indicates that telomere shortening enhances or aggravates lung fibrosis induced by LPS and suggests that telomere shortening confers an inherent promoting factor to pulmonary fibrosis. In addition, we have confirmed the enhancing role of telomere shortening in pulmonary fibrosis in bleomycin-induced pulmonary fibrosis with low dose of 0.3 mg/kg. Bleomycin is a widely used agent to induce pulmonary fibrosis, and that the reason another laboratory previously did not observe the enhancing effect of telomere shortening in bleomycin-induced pulmonary fibrosis may be due to the usage of a high dose of bleomycin [23]. Consistently, it has been shown recently that short telomeres accelerate the pulmonary fibrosis induced by bleomycin with low dose of 0.5 mg/kg [49]. Moreover, we have shown that telomere shortening mice automatically develop limited pulmonary fibrosis, as demonstrated by significant increases in the number of myofibroblasts, α-SMA level and in the deposition of collagen I, but the lack of such a remarkable increase in histopathological score or alveolar thickness suggests that telomere shortening may have already triggered some mediators to promote genesis of pulmonary fibrosis.
Interestingly, in our study, we have shown that telomere shortening activates TGF-β/Smads signaling in lungs as indicated by markedly increased levels of phosphoralated Smad3 and TGF-β1. TGF-β/Smads signaling has multiple functions in different physiological processes including, but not limited to morphogenesis, adult stem cell differentiation, immune regulation,wound healing and inflammation [50, 51]. Once activated, the ligand such as TGF-β1 binds to its receptors in cellular membrane, then Smad2 and Smad3 located in cytoplasm are both phosphorylated, and translocated with Smad4 into nuclei. Over there, phosphorylated Smad2 and Smad3 forms a complex with Smad4, sometime also with other transcription factors, activating transcription of target genes in the downstream of TGF-β/Smads signaling pathway. Therefore, elevated phosphorylated Smad2/3 level is regarded as the activation of TGF-β/Smads signaling pathway. TGF-β/Smads signaling has been demonstrated to contribute to tissue fibrosis process, including kidney fibrosis and lung fibrosis [52,53,54,55]. Moreover, TGF-β1 has shown a function in promoting cellular differentiation from fibroblast to myofibroblast, the latter is regarded as the ‘prime criminal’ of lung fibrosis [56]. Thus, our data suggest telomere shortening enhance LPS or bleomycin-induced lung fibrosis through increased TGF-β/Smads signaling activity. Further, this can also explain why telomere shortening mice developed limited lung fibrosis automatically. The activation of TGF-β/Smads in the lungs of telomere shortening mice may be due to the significantly elevated TGF-β1 levels in these lungs as we observed. However, where does the TGF-β1 come from or what is the detailed mechanism of TGF-β/Smads activation in the lungs of telomere shortening mice, these questions may need further investigation.
LPS is a chemical widely used to induce lung injury, which usually includes destruction of lung integrity, stimulation of inflammatory response and triggering of cell apoptosis [57,58,59]. In our study, we have shown that LPS-treated telomere shortening mouse lungs have a significant increased apoptotic cells than do LPS-treated WT mouse lungs, suggesting that telomere shortening confers genetic susceptibility to LPS challenge in lung injury. The finding that untreated G3 Terc−/− mice have significantly increased apoptotic lung cells than do WT mice suggests that telomere shortening triggers cell apoptosis in the lungs. In lung parenchyma, alveolar epithelia consist of alveolar type I (AT1) cells and AT2 cells; the latter act as stem cells to repair epithelial injuries [36]. Recent studies indicate that AT2 cells are adult stem cells in the lung [37, 38]. Further, we have shown that LPS-treated G3 Terc−/− mouse lungs have a significant increased AT2 apoptotic rate than do LPS-treated WT mouse lungs, suggesting that telomere shortening confers genetic susceptibility to LPS challenge in AT2 cell apoptosis, and stem cells in LPS-treated G3 Terc−/− lungs are significantly compromised. This in turn likely leads to poor capacity for repairing significant increased lung injuries in this group, and contributes the abnormal repairing leading to pulmonary fibrosis.
In our study we have shown that telomere shortening increases LPS-induced cellular senescence in the lungs, which suggests that telomere shortening mice are more susceptible to LPS exposure to generate lung cellular senescence. Previous studies have employed intratracheal LPS instillation to establish animal models of acute lung injury [24,25,26,27,28]. Through toll-like receptor 4 (TLR4) and subsequent NF-κB signaling activation, the release of cytokines and chemokines may recruit inflammatory cells such as neutrophils and macrophages to initiate the inflammation process [60, 61]. Meanwhile, the release of matrix metallopeptidase and reactive oxygen species (ROS) from inflammatory cells may contribute to the degradation of lung architecture, amplifying inflammation and triggering cell apoptosis [57,58,59]. Furthermore, recent studies have indicated that LPS induces cellular senescence in vitro may be via hydrogen peroxide production [34]. Cellular senescence is a state of cell cycle arrest that can be induced by telomere shortening, DNA damage, stress and developmental signals [62, 63]. Senescent cells may lose their proliferation capacity that stem cells always possess [64]. We have consistently shown that high numbers of inflammatory cells such as neutrophils and macrophages occur in LPS-treated lungs. Moreover, the finding that telomere shortening enhances LPS-induced cellular senescence in the lungs suggests that lung cells in LPS-treated G3 Terc−/− lungs are significantly compromised, whose cells may include lung stem cells, which play a role in repairing lung injuries. Moreover, recent studies indicate that senescent cells may be detrimental to organs, and removing of senescent cells in mice lead to tissue rejuvenation [65,66,67]. Our observation that LPS-treated G3 Terc−/− lungs show more-severe cellular senescence than do lungs from any of the other 3 experimental groups may suggest that telomere shortening combined with LPS treatment can trigger the significant loss of lung function.
In this study, we showed that significant inflammatory cell infiltration and NF-κB signaling activation are presented in both LPS- and bleomycin-induced models of pulmonary fibrosis. This finding suggests that inflammation is associated with lung injury or pulmonary fibrosis. Because inflammatory cells were detected in the lungs of patients with IPF, the earliest hypothesis was that inflammation triggered the onset of IPF, but anti-inflammation therapy has subsequently been shown to have little benefit [53, 68]. Thus, the role of inflammation in pulmonary fibrosis is controversial. One study found that lung murine herpesvirus 68 (MHV68) infection produced progressive pulmonary fibrosis in interferon-γ receptor (IFN-γR)−/− mice [69,70,71]. Additionally, a genetic study on a Finnish cohort of familial pulmonary fibrosis identified the pathological gene ELMOD2, whose encoded protein has anti-viral functions [72, 73]. Recent research has posited that pulmonary fibrosis occurs in a two-step process, in which inflammation causes lung injury, then tissue fibrosis develops [74]. We have shown that telomere shortening induces cellular senescence and exacerbates LPS-induced cellular senescence in lungs. It is likely that the cellular senescence contributes to lung inflammatory cell infiltration because senescent cells may secrete bioactive proinflammatory proteins called senescence-associated secretory phenotypes (SASPs) [35, 75]. Interestingly, a recent study indicates that inflammation induces telomere dysfunction and accelerates aging in mice, which suggests that the interaction between inflammation and cell senescence creates a vicious circle for the lungs [76]. Moreover, we have shown that telomere shortening mouse lungs have increased NF-κB signaling; this signaling pathway is well known for its involvement in immunity and inflammatory responses. Previous findings have also indicated that NF-κB activities increased in aging and aging-related chronic diseases and that inhibiting NF-κB delayed cellular senescence and aging in progeroid mice and normal mice, respectively, which suggests that the mechanism by which DNA damage causes aging is partly through NF-κB activation [77]. Since we observe that telomere shortening leads to a remarkable increase in both cellular senescence and NF-κB activity in the lungs, it is possible that telomere shortening can induce cellular senescence partly through NF-κB signaling as well as through the well-known p53–p21 pathway.
In summary, we have shown that telomere shortening activated TGF-β/Smads signaling, this activation is significantly increased in the lungs of LPS- or bleomycin-treated telomere shortening mice. In addition, we have shown that telomere shortening aggravated LPS- or bleomycin-induced pulmonary fibrosis. This tissue fibrosis is accompanied with significant increases of cellular senescence, apoptosis, inflammatory cell infiltration and NF-κB activity. Our data suggest telomere shortening cooperated with harmful environmental exposure accelerates the development of pulmonary fibrosis mainly through creating a pro-fibrosis niche as increased TGF-β/Smads signaling in lungs.
References
King TE Jr., Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961.
Cottin V. Changing the idiopathic pulmonary fibrosis treatment approach and improving patient outcomes. Eur Respir Rev. 2012;21:161–7.
Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:810–6.
Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–26.
Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45:613–20.
Lawson WE, Loyd JE, Degryse AL. Genetics in pulmonary fibrosis-familial cases provide clues to the pathogenesis of idiopathic pulmonary fibrosis. Am J Med Sci. 2011;341:439–43.
Cronkhite JT, Xing C, Raghu G, Chin KM, Torres F, Rosenblatt RL, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:729–37.
Diaz de Leon A, Cronkhite JT, Katzenstein AL, Godwin JD, Raghu G, Glazer CS, et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One. 2010;5:e10680.
Garcia CK. Idiopathic pulmonary fibrosis: update on genetic discoveries. Proc Am Thorac Soc. 2011;8:158–62.
Tsang AR, Wyatt HD, Ting NS, Beattie TL. hTERT mutations associated with idiopathic pulmonary fibrosis affect telomerase activity, telomere length, and cell growth by distinct mechanisms. Aging Cell. 2012;11:482–90.
Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105:13051–6.
Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13:693–704.
Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP, et al. The RNA component of human telomerase. Science. 1995;269:1236–41.
Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–13.
Broccoli D, Young JW, de Lange T. Telomerase activity in normal and malignant hematopoietic cells. Proc Natl Acad Sci U S A. 1995;92:9082–6.
Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18:173–9.
Choi J, Southworth LK, Sarin KY, Venteicher AS, Ma W, Chang W, et al. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet. 2008;4:e10.
Park JI, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M, et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature. 2009;460:66–72.
Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34.
Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, et al. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–12.
Jackson SR, Lee J, Reddy R, Williams GN, Kikuchi A, Freiberg Y, et al. Partial pneumonectomy of telomerase null mice carrying shortened telomeres initiates cell growth arrest resulting in a limited compensatory growth response. Am J Physiol Lung Cell Mol Physiol. 2011;300:L898–909.
Lee J, Reddy R, Barsky L, Scholes J, Chen H, Shi W, et al. Lung alveolar integrity is compromised by telomere shortening in telomerase-null mice. Am J Physiol Lung Cell Mol Physiol. 2009;296:L57–70.
Degryse AL, Xu XC, Newman JL, Mitchell DB, Tanjore H, Polosukhin VV, et al. Telomerase deficiency does not alter bleomycin-induced fibrosis in mice. Exp Lung Res. 2012;38:124–34.
Chen Q, Yang Y, Huang Y, Pan C, Liu L, Qiu H. Angiotensin-(1-7) attenuates lung fibrosis by way of Mas receptor in acute lung injury. J Surg Res. 2013;185:740–47.
Gao Y, Wang N, Liu L, Liu Y, Zhang J. Relationship between mammalian target of rapamycin and autophagy in lipopolysaccharide-induced lung injury. J Surg Res. 2016;201:356–63.
Ma B, Zhou PY, Ni W, Wei W, Ben DF, Lu W, et al. Inhibition of activin receptor-like kinase 5 induces matrix metallo-peptidase 9 expression and aggravates lipopolysaccharide-induced pulmonary injury in mice. Eur Rev Med Pharmacol Sci. 2013;17:1051–59.
Wei W, Ma B, Li HY, Jia Y, Lv K, Wang G, et al. Biphasic effects of selective inhibition of transforming growth factor beta1 activin receptor-like kinase on LPS-induced lung injury. Shock. 2010;33:218–24.
Zhang X, Sun CY, Zhang YB, Guo HZ, Feng XX, Peng SZ, et al. Kegan Liyan oral liquid ameliorates lipopolysaccharide-induced acute lung injury through inhibition of TLR4-mediated NF-kappaB signaling pathway and MMP-9 expression. J Ethnopharmacol. 2016;186:91–102.
Hagiwara S, Iwasaka H, Matsumoto S, Noguchi T, Yoshioka H. Association between heat stress protein 70 induction and decreased pulmonary fibrosis in an animal model of acute lung injury. Lung. 2007;185:287–93.
Hagiwara S, Iwasaka H, Matsumoto S, Noguchi T. Introduction of antisense oligonucleotides to heat shock protein 47 prevents pulmonary fibrosis in lipopolysaccharide-induced pneumopathy of the rat. J Ethnopharmacol. 2007;564:174–80.
Song Z, Zhang J, Ju Z, Rudolph KL. Telomere dysfunctional environment induces loss of quiescence and inherent impairments of hematopoietic stem cell function. Aging Cell. 2012;11:449–55.
Lawson WE, Polosukhin VV, Stathopoulos GT, Zoia O, Han W, Lane KB, et al. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am J Pathol. 2005;167:1267–77.
Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–6.
Kim CO, Huh AJ, Han SH, Kim JM. Analysis of cellular senescence induced by lipopolysaccharide in pulmonary alveolar epithelial cells. Arch Gerontol Geriatr. 2012;54:e35–41.
Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–56.
Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res. 2001;2:33–46.
Alder JK, Barkauskas CE, Limjunyawong N, Stanley SE, Kembou F, Tuder RM, et al. Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci U S A. 2015;112:5099–104.
Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–36.
Stapleton CM, Jaradat M, Dixon D, Kang HS, Kim SC, Liao G, et al. Enhanced susceptibility of staggerer (RORalphasg’sg) mice to lipopolysaccharide-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2005;289:L144–L152.
Suri SS, Mills S, Aulakh GK, Rakotondradany F, Fenniri H, Singh B. RGD-tagged helical rosette nanotubes aggravate acute lipopolysaccharide-induced lung inflammation. Int J Nanomed. 2011;6:3113–23.
Yan Z, Xiaoyu Z, Zhixin S, Di Q, Xinyu D, Jing X, et al. Rapamycin attenuates acute lung injury induced by LPS through inhibition of Th17 cell proliferation in mice. Sci Rep. 2016;6:20156.
Ferretti S, Bonneau O, Dubois GR, Jones CE, Trifilieff AI. L.-17 produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J Immunol. 2003;170:2106–12.
Savov JD, Gavett SH, Brass DM, Costa DL, Schwartz DA. Neutrophils play a critical role in development of LPS-induced airway disease. Am J Physiol Lung Cell Mol Physiol. 2002;283:L952–L962.
Madjdpour C, Jewell UR, Kneller S, Ziegler U, Schwendener R, Booy C, et al. Decreased alveolar oxygen induces lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2003;284:L360–L367.
Thorne PS, McCray PB, Howe TS, O’Neill MA. Early-onset inflammatory responses in vivo to adenoviral vectors in the presence or absence of lipopolysaccharide-induced inflammation. Am J Respir Cell Mol Biol. 1999;20:1155–64.
Chen R, Zhang K, Chen H, Zhao X, Wang J, Li L, et al. Telomerase deficiency causes alveolar stem cell senescence-associated low-grade inflammation in lungs. J Biol Chem. 2015;290:30813–29.
Maguire O, Collins C, O’Loughlin K, Miecznikowski J, Minderman H. Quantifying nuclear p65 as a parameter for NF-kappaB activation: Correlation between ImageStream cytometry, microscopy, and Western blot. Cytometry A. 2011;79:461–9.
Jeyaseelan S, Young SK, Fessler MB, Liu Y, Malcolm KC, Yamamoto M, et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF)-mediated signaling contributes to innate immune responses in the lung during Escherichia coli pneumonia. J Immunol. 2007;178:3153–60.
Povedano JM, Martinez P, Flores JM, Mulero F, Blasco MA. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep. 2015;12:286–99.
Santibanez JF, Quintanilla M, Bernabeu C. TGF-beta/TGF-beta receptor system and its role in physiological and pathological conditions. Clin Sci. 2011;121:233–51.
Lan HY. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int J Biol Sci. 2011;7:1056–67.
Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat Commun. 2012;3:735.
Coward WR, Saini G, Jenkins G. The pathogenesis of idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2010;4:367–88.
Lan HY, Chung AC. TGF-beta/Smad signaling in kidney disease. Semin Nephrol. 2012;32:236–43.
Meng XM, Huang XR, Xiao J, Chung AC, Qin W, Chen HY, et al. Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 2012;81:266–79.
Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007;132:1311–21.
Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol. 2014;35:24–32.
Hoffmann U, Bertsch T, Dvortsak E, Liebetrau C, Lang S, Liebe V, et al. Matrix-metalloproteinases and their inhibitors are elevated in severe sepsis: prognostic value of TIMP-1 in severe sepsis. Scand J Infect Dis. 2006;38:867–72.
Hoshino M, Nakamura Y, Sim J, Shimojo J, Isogai S. Bronchial subepithelial fibrosis and expression of matrix metalloproteinase-9 in asthmatic airway inflammation. J Allergy Clin Immunol. 1998;102:783–8.
Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med. 2012;185:1225–34.
Togbe D, Schnyder-Candrian S, Schnyder B, Doz E, Noulin N, Janot L, et al. Toll-like receptor and tumour necrosis factor dependent endotoxin-induced acute lung injury. Int J Exp Pathol. 2007;88:387–91.
Banito A, Lowe SW. A new development in senescence. Cell. 2013;155:977–8.
Pawlikowski JS, Adams PD, Nelson DM. Senescence at a glance. J Cell Sci. 2013;126:4061–7.
Schultz MB, Sinclair DA. When stem cells grow old: phenotypes and mechanisms of stem cell aging. Development. 2016;143:3–14.
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–9.
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6.
Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83.
Zoz DF, Lawson WE, Blackwell TS. Idiopathic pulmonary fibrosis: a disorder of epithelial cell dysfunction. Am J Med Sci. 2011;341:435–8.
Kropski JA, Lawson WE, Blackwell TS. Right place, right time: the evolving role of herpesvirus infection as a “second hit” in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L441–L444.
Naik PK, Moore BB. Viral infection and aging as cofactors for the development of pulmonary fibrosis. Expert Rev Respir Med. 2010;4:759–71.
Mora AL, Torres-González E, Rojas M, Xu J, Ritzenthaler J, Speck SH, et al. Control of virus reactivation arrests pulmonary herpesvirus-induced fibrosis in IFN-gamma receptor-deficient mice. Am J Respir Crit Care Med. 2007;175:1139–50.
Hodgson U, Pulkkinen V, Dixon M, Peyrard-Janvid M, Rehn M, Lahermo P, et al. ELMOD2 is a candidate gene for familial idiopathic pulmonary fibrosis. Am J Hum Genet. 2006;79:149–54.
Pulkkinen V, Bruce S, Rintahaka J, Hodgson U, Laitinen T, Alenius H, et al. ELMOD2, a candidate gene for idiopathic pulmonary fibrosis, regulates antiviral responses. FASEB J. 2010;24:1167–77.
Cheng T, Liu Q, Zhang R, Zhang Y, Chen J, Yu R, et al. Lysyl oxidase promotes bleomycin-induced lung fibrosis through modulating inflammation. J Mol Cell Biol. 2014;6:506–15.
Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010;16:238–46.
Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun. 2014;2:4172.
Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, et al. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest. 2012;122:2601–12.
Acknowledgements
This work is funded by Hangzhou Normal University Grant PF14002004007 and of Zhejiang Province Natural Science Foundation Grant LY16C070003 to K. Zhang.
Author information
Authors and Affiliations
Contributions
Yi.L., Y.S., and Ya.L. performed experiments; Yi.L. and Y.S. prepared figures; K.Z., Yi.L., Y.S. and X.P. analyzed the data; K.Z. and Yi.L. drafted the manuscript; K.Z., Yi.L. and X.P. edited and revised the manuscript; K.Z. approved the final version of the manuscript; and K.Z., Yi.L., and Y.S. were responsible for the conception and design of the research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Liu, Yy., Shi, Y., Liu, Y. et al. Telomere shortening activates TGF-β/Smads signaling in lungs and enhances both lipopolysaccharide and bleomycin-induced pulmonary fibrosis. Acta Pharmacol Sin 39, 1735–1745 (2018). https://doi.org/10.1038/s41401-018-0007-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0007-9
Keywords
This article is cited by
-
TFEB: a double-edged sword for tumor metastasis
Journal of Molecular Medicine (2023)
-
TGF-β-induced α-SMA expression is mediated by C/EBPβ acetylation in human alveolar epithelial cells
Molecular Medicine (2021)
-
Metformin attenuates TGF-β1-induced pulmonary fibrosis through inhibition of transglutaminase 2 and subsequent TGF-β pathways
3 Biotech (2020)

{kind=link}
{kind=link}
{kind=link}