
Abstract
Liquid-crystalline (LC) Au complexes with siloxane groups at the termini of flexible chains were synthesized. The effects of the molecular and molecular aggregate structures on the luminescence behavior of the complexes were investigated. All complexes used in this study showed LC phases. No direct effect of the siloxane group on the luminescence behavior of the complexes in solution was observed. However, in condensed phases, different luminescence colors were observed depending on the aggregate structure due to the effect of intermolecular interactions. Thus, the luminescence color of the Au complexes can be controlled by the intermolecular interactions based on the structure of the molecular aggregates. The complex with the siloxane group developed in this study showed two different luminescence colors, which can be controlled by changing the aggregate structures induced by the phase transition.
Similar content being viewed by others
Introduction
To be applicable in organic light-emitting devices, luminescent materials must show strong emission in condensed phases [1]. Generally, the luminescence intensity of most organic compounds diminishes significantly in condensed phases because of the concentration-quenching effect or aggregation-induced quenching [1]. However, it was recently reported that aggregation of some types of molecules in their condensed phases can actually enhance emission behavior, resulting in strong photoluminescence. Since luminescence from these materials is induced by aggregation, the process was termed “aggregation-induced emission” (AIE) [2]. The luminescence properties of AIE materials are strongly affected by both the molecular structures of the materials and the structures of their molecular aggregates [3].
We previously reported that Au(I) complexes show AIE properties; while nonaggregated Au complexes in dilute solutions are not emissive, their aggregates are emissive owing to intermolecular interactions, e.g., an interaction between the Au atoms (aurophilic interaction) [4,5,6,7]. It is expected that the luminescence properties can be tuned by modifying the molecular aggregate structure [8,9,10]. It is possible to control the aggregate structure using liquid crystal behavior because liquid crystals exhibit fluidity and orientational order and respond to external stimuli. We recently developed a series of Au complexes with liquid-crystalline (LC) features. Intense luminescence was observed from the crystals prepared with these complexes, and the luminescence could be controlled by adjusting the aggregate structures by a phase transition [11,12,13,14,15]. For instance, complex 1b (Fig. 1) showed a reversible color change between blue and greenish-blue by switching between the crystalline (Cry) and LC phases [16].

Chemical structures of the materials used in this study
The aggregate structures are sensitive to the molecular structure; even when the molecules in the aggregate structures have the same core unit structure, they can show different behaviors due to slight differences in a small part of the molecular structure. We previously reported that a minute change in the structure of a flexible alkyl chain of LC Au complexes induced slight changes in the aggregated crystal structure, resulting in large changes in the luminescence color of the material [15].
In this study, we investigated the effects of the primary molecular structure and the structure of molecular aggregates on the luminescence of Au complexes. We synthesized novel LC Au(I) complexes (2 in Fig. 1) in which a siloxane group was introduced at the terminus of a flexible chain. The siloxane group is flexible and more bulky than an alkoxy chain; thus, it is possible to induce large changes in the aggregate structure in both the Cry and LC phases. The luminescence behavior of the new complexes was compared to the luminescence characteristics of the previously reported compounds with the same core (luminescence center), 1, in order to discuss the effects of molecular and molecular aggregate structures.
Experimental procedures
Materials
Gold complexes 1 and 2 were synthesized from the corresponding 4-alkoxy-4′-isocyanophenyl ligand and (tht)AuCl(tht = tetrahydrothiophene) according to previously reported procedures with some modifications (Scheme 1) [16, 17]. The detailed synthesis procedure for 1 has already been reported [16]. Unless otherwise stated, all solvents and reagents were obtained from commercial suppliers and were used without further purification. Column chromatography was carried out with silica gel (Wakosil® C-200, 64–210 µm), and thin-layer chromatography (TLC) was performed on silica-gel TLC plates (Merck, Silica-gel 60 F254). 1H nuclear magnetic resonance (NMR) and 13C NMR spectra were recorded with a JEOL ECS-400 spectrometer at 400 MHz for 1H and 100 MHz for 13C using the residual 1H and 13C in the NMR solvent as an internal reference. Elemental analyses for C, H, N, and Cl were conducted with a Micro Corder JM10 (J-Science).

Synthesis of Au(I) complexes 2a and 2b (n = 5 and 6). a Potassium carbonate, 18-crown-6, acetone, reflux, 22 h. b Hydrazine monohydrate, 10% Pd/C, ethanol, 2 h, reflux. c Formic acid, toluene, reflux, 4 h. d Pentamethyldisiloxane, Karstedt’s catalyst, dry toluene, 80 °C, 20 h. e Triphosgene, triethyl amine, dichloromethane, rt, 2 h. f (tht)AuCl, dichloromethane, rt, 4 h. The total yields of the complexes were 32% for 2a and 26% for 2b from 4-nitrophenol, respectively
Compound 2a
(tht)AuCl (0.23 g, 0.69 mmol) was added to a dichloromethane solution (8 mL) of 7a (0.28 g, 0.84 mmol), and the mixture was stirred at room temperature for 40 min. The solvent was then removed under reduced pressure, and the residue was purified by recrystallization from a dichloromethane/hexane mixture. Colorless crystals (0.38 g, 0.67 mmol, 95%) were obtained as the target material 2a. 1H NMR (400 MHz, CDCl3, δ): 7.44 (dd, J = 6.9, 2.2 Hz; 2H; 2,6-H in phenyl); 6.95 (dd, J = 6.7, 2.0 Hz; 2H; 3,5-H in phenyl); 3.99 (t, J = 6.5 Hz; 2H; OCH2); 1.81 (quin, J = 7.0 Hz; 2H; OCH2CH2); 1.46 (m; 2H; OCH2CH2CH2); 1.37 (m; 2H, SiCH2CH2); 0.54 (t, J = 8.1 Hz; 2H, SiCH2); 0.06 (s; 9H; (CH3)3SiO); and 0.04 (s; 6H; OSi(CH3)2). 13C NMR (100 MHz, CDCl3, δ): 161.3; 128.5; 115.6; 68.7; 29.5; 28.7; 23.0; 18.22; 1.97; and 0.3. Elemental analysis for C17H29AuClNO2Si2: C, 35.95; H, 5.15; N, 2.47; and Cl, 6.24. Found: C, 35.81; H, 4.74; N, 2.55; and Cl, 6.47.
Compound 2b
Compound 2b was synthesized by following the same procedure as that used for the synthesis of 2a with a yield of 98%. 1H NMR (400 MHz, CDCl3, δ): 7.44 (dd, J = 7.0, 2.2 Hz; 2H; 2,6-H in phenyl); 6.94 (dd, J = 7.0, 2.0 Hz; 2H; 3,5-H in phenyl); 3.99 (t, J = 6.5 Hz; 2H; OCH2); 1.80 (quin, J = 7.0 Hz; 2H; OCH2CH2); 1.46 (m; 2H; OCH2CH2CH2); 1.35 (m; 4H, SiCH2CH2); 0.51 (t, J = 8.1 Hz; 2H, SiCH2); 0.06 (s; 9H; (CH3)3SiO); and 0.04 (s; 6H; OSi(CH3)2). 13C NMR (100 MHz, CDCl3, δ): 161.6; 128.7; 115.2; 68.7; 33.5; 28.3; 23.6; 18.0; 2.5; and 0.3. Elemental analysis for C18H31AuClNO2Si2: C, 37.15; H, 5.37; N, 2.41; and Cl, 6.09. Found: C, 39.93; H, 4.85; N, 2.41; and Cl, 6.47.
X-ray crystallography
Single crystals of the Au complexes 2a and 2b were obtained by slow evaporation from a mixed solvent system (dichloromethane/hexane). The crystallographic data for complexes 1a and 1b have been reported previously [16]. The obtained crystal was mounted on a glass fiber. The omega scanning technique was used to collect the reflection data using a Bruker D8 goniometer with monochromatized MoKα radiation (λ = 0.71075 Å). The initial structure of the unit cell was determined by a direct method using APEX3 software (Bruker). The structural model was refined by the full-matrix least-square method using SHELXL-2014/6 software (Sheldrick, 2014); all additional calculations were performed using this software. Crystal data for the new Au complexes obtained in this study were indexed and included in the Cambridge Crystallographic Centre (CCDC) database with the following reference numbers: 1816839 and 1816838 for 2a and 2b, respectively. The indexed database contains additional supplementary crystallographic data for this paper and may be accessed without charge at http://www.ccdc.cam.ac.uk.conts/retrieving.html.
Phase transition properties
The LC behavior of the complexes was observed using polarizing optical microscopy (POM) with an Olympus BX51 microscope equipped with a hot stage (Instec HC302 hot stage and mk1000 controller) at a heating rate of 5 °C min−1. The thermodynamic properties of the complexes were evaluated using differential scanning calorimetry (DSC; SII X-DSC7000) at a scanning rate of 5 °C min−1. At least three scans were performed to check the reproducibility. To assess the thermochemical stability, thermogravimetry-differential thermal analysis (TG/DTA) was carried out using a Shimadzu DTG-60AH instrument at a heating rate of 5 °C min−1. The interlayer spacing of the Sme phase was determined using X-ray diffractometry (XRD; Ultima IV XRD-DSC IIx, Rigaku) with a D/tex-Ultra detector for the small-angle region and a scintillation counter for the wide-angle region. The temperature was controlled with a built-in DSC unit (Rigaku, ThermoPlus2, DSC8230) at a scanning rate of 10 °C min–1.
Photophysical properties
Ultraviolet-visible absorption and steady-state photoluminescence spectra were recorded on a JASCO V-550 absorption spectrometer and a Hitachi F-7000 fluorescence spectrometer with an R928 photomultiplier (Hamamatsu) detector, respectively. The same crystals prepared for the single-crystal X-ray structure analysis were used for the measurements in the Cry and LC phases. The materials were placed between a pair of quartz plates and set on a homemade heating stage to record the spectra at a controlled temperature. Photoluminescence quantum yields were determined using a calibrated integrating sphere (Hitachi). Photoluminescence lifetimes were measured with a Nd:YAG laser (Continuum, Minilite II: λ = 355 nm; pulse width = 4 ns (full-width-half-maximum); repetition rate = 10 Hz), and decay profiles were recorded with a streak camera (Hamamatsu, C7700).
Results and discussion
Synthesis of Au complexes
New complexes 2a and 2b were successfully synthesized from 4-nitrophenol in acceptable yields; the total yields of the complexes from 4-nitrophenol were 32% for 2a and 26% for 2b. Complexes 2a and 2b were fully characterized by 1H and 13C NMR and elemental analysis. Although the difference between the theoretical and observed data in the elemental analysis for 2b appears to be large, no significant signal of impurities was observed in either the 1H NMR or 13C NMR spectrum (Figures S3 and S4 in the Electronic Supplementary Information (ESI)). Thus, we can conclude that the obtained complexes were fairly pure and that the impurities do not affect the thermodynamic and photophysical properties of the materials.
Crystal structure of Au complexes
Single-crystal X-ray crystallography was performed for 2a and 2b (with siloxane groups) to elucidate the structures of the complexes. The crystal structures of 1a and 1b (without siloxane groups) have already been reported [16]. Key crystallographic data are summarized in Table S1 in the ESI for all complexes. Complex 2a crystallized in the monoclinic space group P21/c with four formula units per unit cell, while complex 2b crystallized in the triclinic space group P−1 with two formula units per unit cell. The Au atoms were linearly coordinated by isocyanide ligands in both 2a and 2b; thus, the core unit of this molecule had a rod-like structure.
The crystal packing structures of the complexes are shown in Fig. 2, and selected interatomic distances between the neighboring molecules are listed in Table 1. The density of the 2b crystal was higher than that of 2a, while the density of 1b was lower than that of 1a (Table S1). Generally, longer alkyl chains decrease the density of a crystal. Hence, these results indicate that complex 2b was packed more densely in the crystal. In the 2a and 2b crystals, the distances between two Au atoms of neighboring molecules were 3.47 and 3.29 Å, respectively. These intermolecular Au–Au distances were significantly shorter than the sum of the van der Waals radii (3.6 Å) of two gold atoms [18], indicating the presence of aurophilic interaction between the neighboring molecules and suggesting that the molecules formed a dimer with an antiparallel orientation. Since the distance between the Au atoms in 2b (3.29 Å) was shorter than those in the other complexes (3.34–3.47 Å), its aurophilic interaction was the strongest. For 2a, the aurophilic interactions expanded, and the molecules were connected along the b-axis of the crystal lattice. As a result, 2a formed a linear-polymer-like structure with noncovalent bonds between each repeating unit in the crystal (Figure S5), similar to the structure of the 1a crystals [16].

Crystal packing structures of a 1a, b 1b, c 2a, and d 2b. Intermolecular Au–Au (orange), Au–π (blue), Au–CH (red), and Cl–CH (black) interactions are indicated by colored lines. Atom colors: gray = C; green = Cl; purple = N; red = O; yellow = Au; pink = Si; light blue = centroid of phenyl ring
It has previously been reported that Au(I) complexes also form aggregates by Au–π interactions, i.e., the interaction between Au atoms and the π-electron system of the aromatic rings [19]. This occurs when the Au–π distance is <4.0 Å and the angle between the vector normal to the aromatic ring and the vector passing through the centroid to the Au atom (α) is ≤20°. In the 2a crystal, the Au atom is located on top of a phenyl ring in the other dimer with a distance of 3.60 Å between the Au atoms and the centroid of the π-phenyl ring, indicating the presence of Au–π interactions. The same intermolecular Au–π interaction was observed in the other crystals. Considering the van der Waals distance of all atoms, we also found several other intermolecular interactions, viz. CH–Cl and CH–Au interactions, as shown in Fig. 2 and Table 1 [20].
Thermodynamics and phase transition behavior
The thermal stability of the complexes was evaluated by TG/DTA. The thermal decomposition temperature was defined as the temperature at which 5% weight loss occurred. It was confirmed that these complexes were thermally stable up to 244 °C for 2a and 261 °C for 2b (Figure S7). The LC behavior of 1 [16] and 2 was investigated by POM and DSC (Figure S8). The phase sequences and phase transition temperatures are shown in Table 2. As shown in Figure S8, no significant difference between the first and second scans in the DSC thermogram was observed. All complexes showed typical fan-shape textures in the POM images; therefore, we concluded that they showed smectic (Sme) LC phases (Fig. 3). Detailed LC phase structural analyses were performed by powder XRD (representative data for 2b are shown in Fig. 4, S9, and S10). Comparing the thermodynamic behaviors of 1 and 2, it can be seen that the siloxane units decreased both the melting point and the clearing point (LC-to-Iso phase transition temperature) of the materials to almost the same extent; as a result, there was no large effect of the siloxane unit on the LC temperature range in these complexes. All complexes showed polymorphism in the Cry phase.

Polarized optical micrographs of a 2a at 136 °C and b 2b at 137 °C. All micrographs were taken during the second cooling process (rate: 5 °C min−1)

a XRD patterns of 2b in various phases; red: Cry1 phase at 25 °C simulated from single-crystal X-ray analysis; green: SmeA phase at 121 °C during heating; purple: SmeA phase at 117 °C during cooling; blue: Cryx phase at 71 °C during cooling. The d spacing (Å) is indicated in brackets. b Structural model of the dimer of 2b formed by Au–Au intermolecular interactions. c, d Schematic illustration of the packing of molecules in the c Cry1 and d SmeA phases
While the molecular structures of complexes 1 and 2 were different from those of the typical rod-like calamitic LC molecules, as mentioned above, the complexes formed dimers with antiparallel orientations in the Cry phase, with the dimers adopting a symmetric rod-like structure [16]. Therefore, we propose that the complexes formed the dimer in the Sme phase, which acted as a mesogen unit forming the Sme phase. Powder XRD results supported the hypothesis that the complexes formed dimers in the LC phases, e.g., in complex 2b, the interlayer spacing of the Sme phase was 35.1 Å (Fig. 4), almost the same as the length of its dimer formed by the aurophilic interaction (35.4 Å, Fig. 4b). We conclude that the mesogen dimers were packed into the Sme layer to form a SmeA phase in 2b, as shown schematically in Fig. 4d; similar behavior was observed for 2a (Figure S9). The Sme phase of 2a was also assigned as an SmeA phase due to its d spacing (30.9 Å) and dimer length (32.4 Å).
Photophysical properties
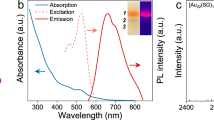
The photophysical properties of 2a and 2b were observed in both the solution and crystal phases. The absorption, photoluminescence, and excitation spectra of the complexes 2a and 2b in dichloromethane solution are shown in Fig. 5. Similar photophysical properties were found for 2a and 2b in solution because both have the same luminescence center. An absorption band was observed at ~270 nm in the solutions. In a dilute solution (10−6 mol L−1), very weak photoluminescence was observed at ~350 nm, and the excitation band appeared at a wavelength close to the absorption band. However, in the higher-concentration solutions (10−5 mol L−1), new luminescence bands appeared at 400–500 nm.

Absorption, normalized photoluminescence, and excitation spectra of complexes in dichloromethane solution and crystals for a 2a and b 2b. Black: absorption spectra in solution (2 × 10−5 mol L−1); green: photoluminescence in solution (λex = 269 nm; solid: 2 × 10−5 mol L−1, dotted: 2 × 10-6 mol L−1); blue: photoluminescence in Cry (λex = 304 nm); red: excitation (solid: in Cry (λem = 428 nm); dotted: in 2 × 10−6 mol L−1 solution (λem = 352 nm for 2a and 360 nm for 2b). (Insets) Absolute intensity of the luminescence spectra of the Cry and solution (2 × 10−5 mol L−1)
Very strong luminescence was observed in the crystals of the complexes; the inset of Fig. 5 compares the absolute intensity of the luminescence spectra in Cry with that in solution. For the 2a crystals, the emission bands appeared only at 400–500 nm with a spectral shape similar to those in 10−5 mol L−1 solutions, while the 2b crystal showed two emission bands at 400–500 nm and at >500 nm. The luminescence band at 400–500 nm matched well with that in solution; however, luminescence above 500 nm was observed only in the crystal. Based on these results, we conclude that the luminescence at 400–500 nm observed in the concentrated solutions and in the crystals was emitted from the aggregates of the molecules, while the luminescence below 400 nm observed only in the dilute solutions was due to monomer emission. Moreover, the materials showed much stronger luminescence in the crystals than in solution; thus, we can also conclude that these materials have AIE characteristics.
The luminescence spectra of 2a and 2b crystals at room temperature are compared in Fig. 6. It can be clearly seen that the luminescence bands above 500 nm appeared only in the 2b crystal (Fig. 6b) and were not observed in complex 1. The Commission Internationale de l’Eclairage (CIE) chromaticity diagram allows for quantitative evaluation of the photoluminescence color, as shown in Fig. 6c for 2a and 2b; the luminescence color of the 2a crystal was deep blue with CIE coordinates of (0.16, 0.06), whereas that of 2b was greenish-blue with CIE coordinates of (0.20, 0.23). The CIE coordinates of the luminescence from complex 1 crystals were the same as those of the 2a crystal.

Photoluminescence spectra of complexes in Cry at room temperature. a Comparison of the luminescence spectra of 2a (red, λex = 304 nm) and 2b (blue, λex = 302 nm). Inset: photographs of crystals taken under UV irradiation at 254 nm. b Comparison of luminescence spectra of 1a (black, λex = 310 nm), 1b (green, λex = 310 nm), 2a (red, λex = 304 nm), and 2b (blue, λex = 302 nm). c CIE chromaticity diagram of the photoluminescence of 2a (red) and 2b (blue) crystals
The luminescence decay profiles were also measured in room temperature crystals for all complexes (Figure S11). The luminescence lifetimes in all complexes were on the order of microseconds, indicating that these emissions were phosphorescence. Complexes 1a, 1b, and 2a showed single exponential decay profiles and had similar lifetimes (1a: 33 µs; 1b: 40 µs; and 2a: 23 µs). However, complex 2b showed a bi-exponential decay with lifetimes of 6 µs (94%) and 12 µs (6%), implying the existence of two excited states corresponding to the luminescence bands at shorter and longer wavelengths, respectively. Luminescence quantum yields (Φ) of the complexes were also estimated for the crystals in air at room temperature. The complexes showed Φ values of 0.04–0.26; 0.18 for 1a, 0.05 for 1b, 0.26 for 2a, and 0.04 for 2b. Generally, it is difficult to observe the phosphorescence at room temperature in air because the long-lived triplet excited state can be easily deactivated by internal motions of the molecule before emission, and the molecular oxygen in air can efficiently quench the triplet excited state; thus, the phosphorescence of the conventional organic molecules can be observed at low temperature, e.g., liquid-nitrogen temperature, under vacuum. However, the complexes used in this study showed intense aggregation-induced phosphorescence in crystals at room temperature in air. This is highly favorable for the use of these crystals as phosphorescence-emitting materials. In these complexes, since the molecules containing the heavy Au atoms were packed densely in the crystals, the internal and external heavy-atom effects work on the molecules to enhance intersystem crossing between the singlet and triplet states. Thus, the triplet excited state was generated very effectively. Moreover, dense packing can restrict the penetration of molecular oxygen into the crystal. These effects resulted in complex crystals with intense phosphorescence at room temperature in air.
Photoluminescence properties in the LC phase
Photoluminescence spectra of 2a and 2b were also recorded at various temperatures in the Cry, Sme, and isotropic (Iso) phases. Figure 7 shows the temperature dependence of the luminescence spectra for 2b as a representative example. All complexes showed photoluminescence in the Sme phase, but the intensity of the luminescence decreased due to the thermal deactivation of the excited state. However, the luminescence disappeared upon transition to the Iso phase during heating. The disappearance of the luminescence was reversible; during cooling, the luminescence was recovered after transition back to the Sme phase. The lack of luminescence in the Iso phase supports our hypothesis that the emission arises from the aggregate (dimer) of the complexes. At the phase transition temperature for the Iso phase, dissociation of the dimer occurs, and the mesogenic dimer vanishes; thus, the Sme-to-Iso phase transition and the disappearance of the luminescence occurred simultaneously.

a, b Temperature dependence of the photoluminescence spectra of 2b (λex = 302 nm) at steps of 10 °C from 40 (orange) to 160 °C (pink): a first heating process and b first cooling process. The inset in b compares normalized luminescence spectra before heating (orange solid), in the Sme phase during the first heating process (blue solid), in the Sme phase during the first cooling process (blue dotted), and in Cry phase during the first cooling process (orange dotted). c, d Luminescence intensity of 2b at 430 (red) and 527 nm (green) as a function of temperature (normalized to the intensity at 40 °C): c during the first heating process and d during the first cooling process
In complex 2a, the luminescence intensity decreased with increasing temperature after the Cry–Sme phase transition, even though the spectral shape was maintained (Figure S12). However, in 2b, an irreversible change in the spectral shape was induced by the phase transition (Fig. 7a). In the first heating process, the intensity of the luminescence peaks above 500 nm decreased monotonically with increasing temperature, whereas the intensity of the peaks at 400–500 nm increased with the Cry–Sme phase transition (Fig. 7c). In the Sme phase, the luminescence bands above 500 nm disappeared, and the spectral shape became similar to that of 2a in the Cry and Sme phases. During cooling from the Iso phase, the luminescence recovered again after the phase transition to the Sme phase; even in the Cry phase, the luminescence bands above 500 nm were not completely recovered, and the Cry peaks showed the same spectral shape as the Sme phase (Fig. 7b, d).
These results showed that complex 2b exhibited distinct luminescence behavior compared to the other complexes. Since luminescence bands above 500 nm were not observed in dilute solution, these bands are thought to have been emitted from the molecular aggregate; therefore, the luminescence behavior depended on both the primary structure of the molecules and the structure of the molecular aggregates. We previously reported that complex 1b showed color sensitivity on the aggregate structure and that the luminescence color can be reversibly controllable via the phase transition between Cryx and Sme phases [16]. As shown in Fig. 4 2b formed a tilted-layer structure in the Cry1 phase, while the mesogenic dimers were not tilted in the Sme phase. These different aggregate structures resulted in the different colors. Furthermore, the molecular packing structure in Cryx obtained by cooling from the Sme phase was also different from that of Cry1, as indicated by the different d001 spacings (Fig. 4), meaning that the tilting angle was different in those crystals. Based on the structural differences, the mode of intermolecular interaction should also differ in these phases. Single-crystal X-ray analysis suggested that Au–Au and Au–π interactions were present in the Cry1 phase of 2b; in addition, the CH–Au interaction was also observed only in this crystal. It is proposed that these intermolecular interactions played a crucial role in the luminescence behavior. We attribute the luminescence bands above 500 nm to the CH–Au interactions; in the Sme phase, these luminescence bands disappeared, meaning that such interactions no longer occurred. After these interactions disappeared when melting the Cry1 phase, they could not be recovered in the Cryx phase (obtained by cooling from the Sme phase); therefore, the same luminescence spectra were observed for both Sme and Cryx phases.
Conclusion
In this study, novel LC Au complexes with siloxane groups at the terminus of the flexible chain were synthesized, and the effects of the molecular and molecular aggregate structures on the luminescence behavior were discussed. Crystals of 2a displayed deep-blue luminescence with bands at 400–500 nm. Since this luminescence band was not observed in a dilute solution and the luminescence intensity was extremely enhanced by the Cry phase, the luminescence showed AIE behavior. The same luminescence spectra were obtained in the room temperature crystals of complex 1; thus, there is no effect of the siloxane group on the luminescence behavior in 2a. On the other hand, in 2b crystals, new luminescence bands were observed above 500 nm and were attributed to CH–Au intermolecular interactions. The CH–Au interactions did not occur after transition to the Sme phase due to structural changes, resulting in the disappearance of the luminescence bands above 500 nm. Consequently, we conclude that the intermolecular interactions played a crucial role in the luminescence behavior and determined the luminescence color of such Au complexes. The luminescence color of the Au complexes can be controlled by modifying the structure of the molecular aggregates and, hence, the intermolecular interactions.
References
Müllen K, Scherf U. Organic light emitting devices: synthesis, properties and applications. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2006.
Luo J, Xie Z, Lam JWY, Cheng L, Chen H, Qiu C, Kwok HS, Zhan X, Liu Y, Zhu D, Tang BZ. Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole. Chem Commun. 2001;1740–1.
Mei J, Leung NLC, Kwok RTK, Lam JWY, Tang BZ. Aggregation-induced emission: together we shine, united we soar! Chem Rev. 2015;115:11718–940.
Bardaji M. In: Laguna A. Modern supramolecular gold chemistry. Weinheim: Liquid crystal, Wiley-VCH Verlag GmbH & Co; 2009. p. 403–28.
Seki T, Tokodai N, Omagari S, Nakanishi T, Hasegawa Y, Iwasa T, Taketsugu T, Ito H. Luminescent mechanochromic 9-anthryl gold(I) isocyanide complex with an emission maximum at 900 nm after mechanical stimulation. J Am Chem Soc. 2017;139:6514–7.
Jin M, Seki T, Ito H. Luminescent mechanochromism of a chiral complex: distinct crystal structures and color changes of racemic and homochiral gold(I) isocyanide complexes with a binaphthyl moiety. Chem Commun. 2016;52:8083–6.
Jin M, Seki T, Ito H. Mechano-responsive luminescence via crystal-to-crystal phase transitions between chiral and non-chiral space groups. J Am Chem Soc. 2017;139:7452–5.
Fujisawa K, Yamada S, Yanagi Y, Yoshioka Y, Kiyohara A, Tsutsumi O. Tuning the photoluminescence of condensed-phase cyclic trinuclear Au(I) complexes through control of their aggregated structures by external stimuli. Sci Rep. 2015;5:7934.
Yamada S, Rokusha Y, Kawano R, Fujisawa K, Tsutsumi O. Mesogenic gold complexes showing aggregation-induced enhancement of phosphorescence in both crystalline and liquid-crystalline phases. Faraday Discuss. 2017;196:269–83.
Yamada S, Yamaguchi S, Tsutsumi O. Electron-density distribution tuning for enhanced thermal stability of luminescent gold complexes. J Mater Chem C. 2017;5:7977–84.
Fujisawa K, Izumi Y, Nagamatsu A, Uno K, Tsutsumi O. Liquid-crystalline behavior and photoluminescence properties of gold(I) complex: relationship between aggregation structure and properties. Mol Cryst Liq Cryst. 2012;563:50–7.
Fujisawa K, Kawakami N, Onishi Y, Izumi Y, Tamai S, Sugimoto N, Tsutsumi O. Photoluminescent properties of liquid crystalline gold(I) isocyanide complexes with a rod-like molecular structure. J Mater Chem C. 2013;1:5359–66.
Sugimoto N, Tamai S, Fujisawa K, Tsutsumi O. Photoluminescent gold(I) complex with biphenyl acetylene ligand showing stable nematic liquid-crystalline phase. Mol Cryst Liq Cryst. 2014;601:97–106.
Younis O, Rokusha Y, Sugimoto N, Fujisawa K, Yamada S, Tsutsumi O. Effects of molecular structure and aggregated structure on photoluminescence properties of liquid-crystalline gold(I) complexes with various aromatic rings. Mol Cryst Liq Cryst. 2015;617:21–31.
Kawano R, Younis O, Ando A, Rokusha Y, Yamada S, Tsutsumi O. Photoluminescence from Au(I) complexes exhibiting color sensitivity to the structure of the molecular aggregates. Chem Lett. 2016;45:66–8.
Fujisawa K, Okuda Y, Izumi Y, Nagamatsu A, Rokusha Y, Sadaike Y, Tsutsumi O. Reversible thermal-mode control of luminescence from liquid-crystalline gold(I) complexes. J Mater Chem C. 2014;2:3549–55.
Usón R, Laguna A, Laguna M. (Tetrahydrothiophene)gold(I) or gold(III) complexes. Inorg Synth. 1989;26:85–91.
White-Morris RL, Olmstead MM, Balch AL. Aurophilic interactions in cationic gold complexes with two isocyanide ligands. Polymorphic yellow and colorless forms of [(cyclohexyl isocyanide)2Au(I)](PF6) with distinct luminescence. J Am Chem Soc. 2003;125:1033–40.
Tiekink ERT, Zukerman SJ. Gold•••π aryl interactions as supramolecular synthons. CrystEngComm. 2009;11:1176–86.
Bakar MA, Sugiuchi M, Iwasaki M, Shichibu Y, Konishi K. Hydrogen bonds to Au atoms in coordinated gold clusters. Nat Commum. 2017;8:576.
Bondi A. van der Waals volumes and radii. J Phys Chem. 1964;68:441–51.
Acknowledgements
This research was supported by the JSPS KAKENSHI (15K05613), the JST Matching Planner Program (MP27115663415), and the Cooperative Research Program of the Network Joint Research Center for Materials and Devices (Tokyo Institute of Technology).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
41428_2018_60_MOESM1_ESM.pdf
Electronic Supplementary Information for Photoluminescence Behavior of Liquid-Crystalline Gold(I) Complexes with Siloxane Group Controlled by Molecular Aggregated Structures in Condensed Phases
Rights and permissions
About this article

Cite this article
Fujisawa, K., Mitsuhashi, F., Anukul, P. et al. Photoluminescence behavior of liquid-crystalline gold(I) complexes with a siloxane group controlled by molecular aggregate structures in condensed phases. Polym J 50, 761–769 (2018). https://doi.org/10.1038/s41428-018-0060-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-018-0060-8
