
Abstract
A new enzyme catalyst consisting of pyrenecarboxaldehyde (PCA) and glucose oxidase (GOx) immobilized on polyethyleneimine (PEI) and a carbon nanotube supporter (CNT/PEI/[PCA/GOx]) is suggested, and the performance and stability of an enzymatic biofuel cell (EBC) using the new catalyst are evaluated. Using PCA, the amount of immobilized GOx increases (3.3 U mg−1) and the electron transfer rate constant of the CNT/PEI/[PCA/GOx] is promoted (11.51 s−1). Also, the catalyst induces excellent EBC performance (maximum power density (MPD) of 2.1 mW cm−2), long-lasting stability (maintenance of 93% of the initial MPD after 4 weeks) and superior catalytic activity (flavin adenine dinucleotide redox reaction rate of 0.62 mA cm−2 and Michaelis–Menten constant of 0.99 mM). These characteristics are ascribed to effects of (i) electron collection due to hydrophobic interactions, (ii) electron transfer pathways due to π-conjugated bonds and (iii) enzyme stabilization due to π-hydrogen bonds that are newly induced by the PCA/GOx composite. The existence of such positive interactions is properly verified using X-ray photoelectron spectroscopy and enzyme activity measurements.
Similar content being viewed by others

Introduction
The demand for clean electrical energy is increasing as a result of weather changes caused by the greenhouse effect and depletion of fossil fuels.1 To meet this demand, related research and development efforts are being eagerly pursued. As one of the efforts, enzymatic biofuel cells (EBCs) that convert bioenergy into electrical energy are being considered.2, 3 In particular, EBC systems employing glucose oxidase (GOx) as a biocatalyst have emerged because of their advantageous properties, such as biocompatibility, excellent selectivity toward specific substrates and strong activity near neutral pH and room temperature.2 In addition, because EBC systems use human body-friendly fuels, such as glucose, glycerol, water and oxygen, for electricity generation, they can be embedded as power generators for the operation of artificial internal organs, such as artificial hearts and brains, insulin pumps or bone stimulators.4, 5, 6, 7, 8
However, in spite of such promising facets of EBC systems, the commercialization of EBCs using GOx has been limited because of their low EBC performance and poor durability.9 The disadvantages are attributed to the low immobilization ratio of GOx molecules, slow GOx-related reaction rate, low GOx activity and easy GOx denaturation. Of them, low GOx immobilization has been considered the main problem.
To increase the amount of immobilized GOx, a variety of methods including non-covalent adsorption, encapsulation, covalent coupling, affinity bonding, entrapment and enzyme crosslinking have been suggested.10, 11, 12, 13, 14, 15 In spite of these attempts, a standard process for GOx immobilization has not yet been developed. Furthermore, GOx immobilization research to date has mostly focused on only increasing the amount of immobilized GOx without serious consideration of how to improve the electron transfer between the immobilized GOx molecules and supporter materials. To enhance the electron transfer, the following materials are currently being studied: (i) electron conductors such as metal nanoparticles, (ii) carbon nanotubes (CNTs) as supporter materials and (iii) conductive polymers as entrapment agents.16, 17 However, because the active sites (flavin adenine dinucleotides (FADs)) of GOx are located deep inside shells consisting of proteins that prevent electron transfer, the conventional immobilization methods are limited, and it is difficult to improve the electron transfer in GOx-based catalytic structures.18
To achieve the facile electron transfer and immobilize large amounts of GOx, this study suggests adopting a new material: pyrenecarboxaldehyde (PCA). The PCA modifies surfaces of the GOx molecules, producing PCA/GOx composites. As support materials, grafting structures of both CNTs that exhibit superior electron transfer capabilities and polyethyleneimine (PEI) as a conductive polymer with excellent biocompatibility are combined (CNT/PEI). In turn, the PCA/GOx composite are directly linked to the CNT/conductive polymer supporter.
To immobilize the GOx molecules, in this study, we propose three main bonding mechanisms. First, active sites (FADs) of the GOx molecules are surrounded by hydrophobic pockets that are physically entrapped with pyrene groups of the PCA via hydrophobic interactions (an electron collection effect).19 Second, aldehyde groups belonging to FAD and PCA are chemically bonded to the free amine groups of the CNT/PEI supporter via a Schiff base formation reaction to form C=N bonds, forming π-conjugated electron transfer pathways (an electron transfer pathway effect). Third, pyrene groups attached to the GOx form π-hydrogen bonds with the free amine groups of PEI. Owing to the π-hydrogen and C=N bonds, the GOx molecules are not denaturized and the corresponding enzyme structures show long-lasting stability and excellent catalytic activity.
To verify effects of using PCA, different types of enzyme catalytic structures (GOx, PCA/GOx, CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx]) are prepared, and their chemical structures are analyzed using ultraviolet–visible spectroscopy and X-ray photoelectron spectroscopy (XPS). To evaluate the electrochemical performances and stability of the structures, cyclic voltammogram (CV) and EBC polarization curves are measured.
Materials and methods
Materials
Multiwall carbon nanotubes (MWCNTs) (with a purity higher than 90%) were obtained from NanoLab (Brington, MA, USA). PCA, GOx (from Aspergillus niger type X-S, 150 000 U g−1 solid), horseradish peroxidase (146 U mg−1 solid), o-dianisidine and PEI solution were purchased from Sigma-Aldrich (Milwaukee, WI, USA).
Fabrication of the enzyme-based catalysts
The CNT/PEI/GOx catalyst was fabricated by alternating deposition using PEI (positive charge) and GOx (negative charge) on the CNTs. Initially, 25 mg of the MWCNTs were dissolved in 5 ml of PEI (2.5% w w−1 in deionized water), then the mixture was sonicated for 1 h and centrifuged at 14 000 r.p.m. for 7 min. The excess PEI was removed using deionized water. Then, the CNT/PEI mixture was immersed in a GOx solution (5 mg ml−1 in 0.01 M phosphate-buffered saline (PBS) (pH 7.4)) for 1 h to complete the CNT/PEI/GOx catalyst. To fabricate the CNT/PCA/GOx catalyst, PCA/GOx was initially synthesized. The PCA/GOx composite that was mixed with 5 mg of GOx and 2 mg of PCA was dissolved in 1 ml of ethanol and then mixed for 2 h. In turn, the PCA/GOx composite was immersed in 5 mg of CNT for 2 h. The mixture was centrifuged at 14 000 r.p.m. for 7 min, and its supernatant was filtered and removed, completing the fabrication of the catalyst. To fabricate the CNT/PEI/PCA/GOx catalyst, the PCA/GOx composite was mixed with CNT/PEI for 2 h. Then, the CNT/PEI/PCA/GOx catalyst was centrifuged at 14 000 r.p.m. for 7 min. All catalysts were stored in a 0.01 M PBS (pH 7.4) solution at 4 °C when not in use.
Chemical characterization of the enzyme-based catalysts
To confirm the bonding mechanism, the chemical structures of the GOx/PCA composites and the corresponding catalytic structures were measured using XPS (Perkin-Elmer PHI 5800 ESCA; Waltham, MA, USA). The XPS system was operated with an Al K monochromatic X-ray source. The binding energy scan range was 280 to 290 eV.
Electrochemical characterization of the enzyme-based catalysts
A computer-connected potentiostat (Bio-Logic SP-240; Bio-Logic, Seyssinet-Pariset, France) was used for the electrochemical measurements. For measuring the CVs, Pt wire and Ag/AgCl (soaked in 3.0 M KCl) served as the counter and reference electrodes, respectively, while the catalysts were loaded on glass carbon electrodes to act as the working electrode. To load the catalysts, catalytic powder was mixed with 1 ml of H2O, and then 10 μm of the catalytic ink was dropped on the glass carbon electrode. The catalytic ink-loaded working electrode was then dried for 45 min. After drying, a 5 wt% Nafion solution was coated on the working electrode to complete the configuration of the working electrode. For the electrolyte, 1 M PBS (pH 7.4) was used, while N2 and air were fed to create specific atmospheres, such as a N2 state and air state.
To measure the polarization curves from the EBC single cell, a potentiostat was also connected with a frequency response analyzer. By coupling the frequency response analyzer with the potentiostat, the power output was analyzed as a product of the current and potential. For the cathode, 100 cm3 min−1 O2 gas was fed to the electrode, while a 0.2 M glucose solution was circulated as fuel for the anode electrode.
Results and discussion
Chemical structures of the catalysts including PCA and PEI
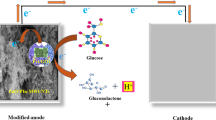
The effects of PCA on three catalytic structures including CNT, PEI and GOx were initially investigated. Figure 1a shows schematic illustrations of the three structures: CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx]. In CNT/PEI/GOx, CNT physically interacted with PEI and GOx, whereas in CNT/[PCA/GOx], PCA was attached to GOx (PCA/GOx composite) to form π–π interactions with CNT. In CNT/PEI/[PCA/GOx], the PCA/GOx composite interacted with CNT/PEI to form C=N and π-hydrogen bonds.

Schematic illustrations showing (a) fabrication of the CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] structures and (b) two different bonds formed by using the PCA/GOx composite.
More specifically, when PCA was attached to GOx molecules to form the PCA/GOx composite, two kinds of bonds between PCA and GOx were expected: (i) pyrene−C=N−GOx bonds between the aldehyde groups of PCA and amine groups of lysine residue on the GOx surface to form a Schiff base formation reaction and (ii) hydrophobic bonds between the pyrene groups belonging to the hydrophobic areas of PCA and hydrophobic sites near the FADs within GOx to form hydrophobic interaction bonds.20 The possible bonds are illustrated in Figure 1b.
The reaction mechanisms of the CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] structures are explained as follows: in CNT/[PCA/GOx], the pyrene groups of the PCA/GOx composite formed π–π interactions with CNT, producing π–π stacking.21, 22 In contrast, in CNT/PEI/[PCA/GOx], the PCA/GOx composite is preferentially immobilized because the (–) charged pyrene rings of the PCA/GOx composite are electrostatically attracted to the (+) charged CNT/PEI. The aldehyde groups of the PCA/GOx composite are then attached to the amine groups of PEI on CNTs to produce C=N bonds. Because the C=N bonds have a high bonding energy (615 kJ mol−1), the bonding energy of CNT/PEI/[PCA/GOx] was also high.23 Moreover, owing to hydrophobic sites near the FAD, electrons generated/consumed by the FAD redox reaction were captured by the pyrene groups of PCA and transferred easily to the CNTs due to π-conjugated electron transfer pathways (FAD–pyrene–C=N–PEI). In addition, the free pyrene groups of PCA that were attached on the surface of GOx interacted with the amine groups of PEI to form π-hydrogen bonds, and PEI could also be physically entrapped in the PCA/GOx composite. The new π-hydrogen bonds and physical entrapment played a critical role in preventing denaturation of the PCA/GOx composite from the CNT/PEI. The possible reaction mechanisms are illustrated in Figure 2.

Schematic illustrations showing electrostatic attractions between the PCA/GOx composite and CNT/PEI and the bonding mechanism of the CNT/PEI/[PCA/GOx] structure.
Unlike the other structures, in CNT/PEI/GOx, dipole interactions between the (+) and (–) charges of PEI and GOx were a major bonding resource.24 Since the dipole interactions are weaker bonding than π–π interactions and C=N double bonds, the dipole bonding energy may have been weaker than that of the other bonds.
To further investigate how well PCA captured GOx molecules to reduce their denaturation, the amount of immobilized GOx was measured using a colorimetric method with a ultraviolet–visible spectrophotometer. The detailed procedure for measuring the activity is explained in the Supplementary Information. According to the data (Supplementary Figure S1), the amount of GOx immobilized in CNT/PEI/[PCA/GOx] was highest, demonstrating that this structure was most effective for increasing the amount of immobilized GOx (Supplementary Figure S1a). From quantitative calculations, % of GOx immobilized in the CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] was 66%, 54% and 42%, respectively (Supplementary Figure S1b and Supplementary Table S1).
To verify such explanations of the bonding mechanisms, chemical structures of the GOx/PCA composite included catalysts were estimated using XPS, and the results are presented in Figures 3 and 4. As shown in Figure 3, GOx molecules were mostly occupied by C-C (sp3) bonds, although small amounts of C=C (sp2) and C-N (C=N) bonds were also observed. In contrast, in the GOx/PCA composite, the peaks for the C=C (sp2) and C-N (C=N) bonds rapidly increased because the newly employed PCA was attached to the amine groups of lysine residues of the GOx surface to form C=N bonds. After attachment, main chemical structure of the GOx/PCA composite was changed from C-C (sp3) bonds to C=C (sp2) bonds. In turn, because of the formation of the C=C bonds, (i) π–π interactions formed between the C=C bonds and the CNTs of CNT/[PCA/GOx] and (ii) π-hydrogen interactions formed between the C=C bonds and the CNT/PEI groups of CNT/PEI/[PCA/GOx].

C1s peaks of the (a) GOx and (b) PCA/GOx structures measured using XPS. C=C (sp2), C-C (sp3), C-N, C=N, C-O and C=O were identified from the C1s peak analysis.

C1s peaks of the (a) CNT/PEI/GOx, (b) CNT/[PCA/GOx] and (c) CNT/PEI/[PCA/GOx] structures measured using XPS. C=C (sp2), C-C (sp3), C-N, C=N, C-O and C=O were identified from the C1s peak analysis.
Meanwhile, small amounts of C=O bonds were observed in the GOx/PCA composite, which were not observed in the GOx molecule. The presence of C=O bonds are a clear evidence of the hydrophobic interactions because the hydrophobic bonds are supposed to be configured by the C=O bonds. Such hydrophobic interactions were observed even in CNT/[PCA/GOx].
Taken together, effects of PCA, as evaluated using XPS measurements, could be summarized in the following two ways: first, most of the PCA were participated in the formation of ‘pyrene–C=N–GOx’ bonds on the surface of GOx due to Schiff base formation reactions, and second, the remaining PCA induced ‘O=C–pyrene–GOx’ bonds near the FAD groups due to hydrophobic interactions. The hydrophobic interactions were confirmed from observations of the pyrene rings and aldehyde groups on the surface of GOx.
The XPS C1s peaks of each catalyst are represented in Figure 4. For CNT/PEI/GOx, the peak of the C-C (sp3) bonds was highest, while a peak for the C-N (C=N) bonds was also observed. When compared with the GOx peak, this result meant that the peak of the C=C (sp2) bonds was considerably reduced, while that of the C-N(C=N) bonds increased. It is attributed to weak physical absorption (dipole interactions) between PEI and GOx. Owing to the weak interactions, the amount of GOx that could be immobilized was limited, and instead, PEI mostly occupied surface of the structure.
The CNT/[PCA/GOx] contained many C=C (sp2) bonds, even more than the GOx/PCA composite. This result was ascribed to the C=C (sp2) bonds of the CNTs. Another important fact was that the amount of C=O bonds increased due to the remaining nonreacted aldehyde groups within the GOx/PCA composite.
Unlike CNT/PEI/GOx and CNT/[PCA/GOx], the amount of C-C (sp2) bonds in CNT/PEI/[PCA/GOx] was relatively high, even though PEI was present. Additionally, the amounts of the C-C (sp2), C-C (sp3) and C-N bonds were very similar to those of the GOx/PCA composite, indicating that the surface of CNT/PEI/[PCA/GOx] was mostly occupied by the GOx/PCA composite. Hence, the free aldehyde groups of PCA chemically reacted with the free amine groups of PEI (hydrophobic interactions), and the chemical bonding promoted immobilization of the GOx molecules. These explanations are well matched with the enzyme activity measurements (Supplementary Figure S1).
Electrochemical characterizations of catalysts including PCA and PEI
It was critical to estimate the role of PCA by measuring catalytic activity of the redox reaction of FAD within GOx: ((GOx (FAD)+2H++2e−↔GOx (FADH2)). Therefore, the CV curves of CNT/GOx and CNT/[PCA/GOx] were measured and compared (Figure 5a). There were two main noticeable results. First, the FAD redox reaction peak of CNT/[PCA/GOx] was far higher than that of CNT/GOx, indicating that PCA had an important role in promoting the FAD redox reaction. Second, the envelope current of CNT/[PCA/GOx] was significantly higher than that of CNT/GOx, indicating that PCA also affected the increase in active surface area.

Cyclic voltammograms of the (a) CNT/GOx and CNT/[PCA/GOx] and (b) CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] structures. For the tests, 1.0 M PBS (pH 7.4) was used as the electrolyte for the N2 state, and the potential scan rate was 100 mV s−1.
To further evaluate effects of the PCA/GOx composites on the catalytic activity of the FAD redox reaction, CV curves of CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] were measured (Figure 5b). According to the figure, the FAD redox reaction peak of CNT/PEI/[PCA/GOx] was highest. From a quantitative analysis of the FAD redox reaction peaks, oxidative peak current densities (the difference between the peak current density and envelope current density (the upper peak differences of the CV curves, ΔJp)) of CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] were 0.1, 0.28 and 0.62 mA cm−2, demonstrating that the FAD reaction increased by ∼3–6-fold because of the use of PCA. Additionally, because envelope currents of the structures were almost similar, it was speculated that the enhanced FAD reaction was due to improvements in the electron transfer.
The CV data of Figure 5 can be compared with the activity measurements of Supplementary Fig. S1 to estimate utilization of the GOx molecules in the catalytic structures. According to the GOx activity measurements, the amounts of GOx immobilized in CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] were 2.1, 2.7 and 3.3 U mg−1 (Supplementary Table S1), respectively, meaning that 1.6-fold more GOx molecules were immobilized on CNT/PEI/[PCA/GOx] than CNT/PEI/GOx. However, the difference in the FAD reaction rates between the two structures, as indicated by ΔJp, was ∼6-fold (the ΔJp values of CNT/PEI/GOx and CNT/PEI/[PCA/GOx] were 0.1 and 0.62 mA cm−2, respectively). It implied that although the amount of GOx immobilized on CNT/PEI/[PCA/GOx] did not rapidly increase compared with CNT/PEI/GOx, the electron transfer (FAD reaction rate) increased four times because of the surface modification of GOx by PCA. Such an increase in the electron transfer of CNT/PEI/[PCA/GOx] was attributed to the (i) electron collection effect of the pyrene rings near the FAD groups and (ii) electron transfer pathway effect due to the π-conjugated C=N bonds.
Next, the catalytic activities of the structures incorporated with PCA (CNT/PEI/[PCA/GOx] and CNT/[PCA/GOx]) were compared. According to the enzyme activity comparison, CNT/PEI/[PCA/GOx] showed 1.3-fold more activity than CNT/[PCA/GOx], while the difference in the FAD reaction rate was almost 2-fold between the two structures. These results imply that changes in the chemical structure from PCA/GOx to PEI/[PCA/GOx] induced approximately twice better electron transfer (FAD reaction rate).
Indeed, the above results indicate that CNT/PEI/[PCA/GOx] exhibited advantageous properties, such as excellent catalytic activity and large amounts of immobilized GOx, and these properties were attributed to the electron collection and electron transfer pathway effects. Namely, the pyrene ring groups of PCA were well attached to the hydrophobic sites near the FADs. Then, the hydrophobic interactions captured the electrons produced/consumed from the FADs. Simultaneously, the C=N bonds produced between the aldehyde groups of PCA and amine groups of PEI or GOx formed π-conjugated electron transfer pathways. These interactions promoted further electron transfer.
To further investigate the electron transfer pathway effect due to PCA (π-conjugated electron transfer pathways induced by C=N bonds between the aldehyde groups of PCA and amine groups of PEI or GOx), the catalytic activities of CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] were compared with those of CNT/[pyrene/GOx] and CNT/PEI/[pyrene/GOx] after measuring their CV curves (Supplementary Figures S2a and b). Here, ‘pyrene’ is a simple pyrene that does not contain aldehyde groups. According to Supplementary Figures S2a and b, the catalytic activities of the FAD redox reactions of CNT/[pyrene/GOx] and CNT/PEI/[Pyrene/GOx] were 0.14 and 0.59 times lower than those of CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx], respectively, proving that the C=N bonds promoted the FAD redox reactions due to the formation of the π-conjugated electron transfer pathways.
In CNT/PEI/[PCA/GOx], the FAD redox reaction peak potential was more positively shifted than that of CNT/PEI/GOx and CNT/[PCA/GOx]. In our hypothesis, this shift was due to deprotonation during the Schiff base formation reaction (the reaction between the aldehyde groups of PCA and amine groups of PEI). Namely, the imine groups produced by the reaction released H+ into the environment and pH values of the catalytic structures decreased.20, 25 To prove this phenomenon, we measured local pH of the three catalysts and confirmed that the local pH of CNT/PEI/[PCA/GOx] shifted negatively (the local pH values of CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] were 7.52, 7.88 and 7.05, respectively). These data are also included in the Supplementary Information (Supplementary Figure S3).
The rate-determining step of all the catalytic structures was also evaluated. For that purpose, relations between the peak current densities and potential scan rates of the structures were measured under various scan rate conditions (Supplementary Figures S4a–f). From the experiments, the FAD redox peaks linearly increased with the potential scan rate, confirming that all the catalytic structures were controlled by surface reactions. Moreover, the ΔEp values of CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] were 30, 15, and 18 mV at a scan rate of 100 mV s−1, whereas the Ipa/Ipc ratio was close to one, demonstrating that the structures were within the quasireversible reaction region.26
The electron transfer rate constant (ks) of the catalytic structures was measured because ks was proportional to the reaction rate, followed by the EBC performance. For measuring the ks, Laviron’s formula was used,24, 27, 28 and the results are represented in Supplementary Figure S5. The ks values of CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] were 10.35±0.32, 11.04±0.41 and 11.51±1.38 s−1, respectively. This trend supports the other chemical characterization results from ultraviolet–visible spectroscopy and XPS.
Based on the above data, the glucose oxidation reactivity of the catalytic structures (the reaction rate of the glucose oxidation reaction) was evaluated because the glucose oxidation reactivity is one of the major factors for determining catalytic activity and EBC performance. To achieve this, effects under two ambient conditions (air state and N2 state) on the catalytic activity of the structures were initially investigated without glucose (Supplementary Figure S6), and then effects of the ambient conditions on the glucose oxidation reactivity of the catalytic structures were measured (Supplementary Figures S7 and S8). According to Supplementary Figures S6a–c, in the air state, the CV curves were downshifted. This downshift was attributed to the reduction of O2 (O2+2H++2e−→H2O2).29, 30 Supplementary Figures S7a–c represent the CV curves of the catalytic structures measured in N2 state with 10 mM glucose. In Supplementary Figure S7, the CV curves remained unchanged irrespective of the glucose concentration. This result indicates that electrons generated by the glucose oxidation reactions (Glucose→2H++2e−+gluconolactone) did not affect the redox reaction of FAD (GOx (FAD)+2H++2e−↔GOx (FADH2)) without a mediator, while a mediator should be considered for transferring electrons between glucose and FAD.31
Supplementary Figures S8a–c present CV curves showing effect of O2 on the glucose oxidation reactivity of the catalytic structures. During the tests, 0.1–10 mM glucose was fed in air state (with O2). Before the addition of glucose, the overall CV pattern was downshifted compared with that in Supplementary Figure S8 with provision for O2 because of the peak overlap due to concurrency of the reduction reactions of O2 (increase in the cathodic current) and redox reaction of FAD (redox reaction peaks observed at −0.47 V vs Ag/AgCl).24 Subsequently, with the increase in the glucose concentration, the overall CV pattern was upshifted, indicating that the cathodic current was reduced (the reduction reaction of O2 decreased). This result was ascribed to the mediating role of O2 (GOx (FADH2)+O2→GOx (FAD)+H2O2), and hence the downshifted CV pattern occurred before provision for glucose could be upshifted with a gradual increase in the glucose concentration.
The apparent Michaelis–Menten constant (Km), maximum current density (Jmax) and glucose sensitivity were also measured using Michaelis–Menten and Lineweaver–Burk plots (Figures 6a and b). The glucose sensitivities of CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] were 12, 15 and 19 μA mM−1 cm−2, respectively, while their Km and Jmax values were 1.11, 1.06 and 0.99 mM and 0.011, 0.013 and 0.017 mA cm−2, respectively. These results were compatible with the other data. Affordability for the FAD redox reaction of the catalytic structures (two-electron and two-proton redox reaction) was also measured using the correlations between the redox peak potentials of FAD and electrolyte pH (Supplementary Figures S9a–c). The redox peak potentials of CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] linearly decreased with slopes of 59, 56 and 51 mV pH−1 for the anode and 59, 58 and 50 mV pH−1 for the cathode, respectively, when the pH increased from 3.0 to 9.0. These slopes were compatible with the theoretical slopes (−58.6 mV pH−1) for the two-electron and two-proton redox reactions, indicating that the FADs of these catalyst structures participated in the desirable two-electron and two-proton redox reactions.32

(a) The relationship between glucose concentration and difference in the reduction peak currents of the CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] structures and (b) their Lineweaver–Burk plots. For the CV tests, 1.0 M PBS (pH 7.4) was used as the electrolyte for the air state, 0–10.0 mM glucose was supplied and the potential scan rate was 100 mV s−1.
Performances and stability of the EBCs using the catalysts including PCA and PEI
We investigated the performances and stability of the EBCs using CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] as anodic catalysts by measuring their polarization curves. Additionally, we evaluated the effects of enzyme catalyst and glucose fuel on the EBC performance. To inspect whether the results were reproducible, polarization curves were measured three times using three samples per catalyst. Figure 7 presents performances of the EBCs. According to Figure 7a, maximum power density (MPD) of the EBC with CNT/PEI/[PCA/GOx] was better than that of the EBCs with other structures (the MPDs of the EBCs using CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] were 1.13±0.054, 1.34±0.093 and 2.19±0.199 mW cm−2, respectively). This result agreed well with Jmax of Figure 6. Namely, the Jmax ratios of CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] were approximately the same as the MPD ratios of the corresponding EBCs. Because power density is proportional to current density, this correlation indicates that the MPD (Figure 7) and Jmax (Figure 6) data are reasonable. Furthermore, our EBC performance was better than that of other similar EBCs, as shown in Table 1.

Polarization curves of (a) EBCs using CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] as the anodic catalysts and Pt/C as the cathodic catalyst and (b) EBCs using CNT/PEI/[PCA/GOx] as the anodic catalyst and CNT/PEI/PCA/Lac as the cathodic catalyst. In the tests, a 0.2 M glucose solution was provided and circulated from an external bottle to the anode chamber of EBC at a rate of 0.1 ml min−1. For the cathodic reaction, 100 cm3 min−1 O2 gas was supplied.
Figure 7b presents polarization curves showing effects of PEI/PCA composite, enzyme catalyst and glucose fuel on the EBC performance. For evaluating the enzyme catalyst effect, the polarization curve of EBC using CNT/[PEI/PCA] catalysts without GOx and laccase molecules was compared with that of the EBC using CNT/PEI/[PCA/GOx] (anode) and CNT/PEI/[PCA/Laccase] (cathode) catalysts without glucose. According to their MPDs, the latter was better than the former by a factor of three (MPDs of the former and latter were 0.158±0.01 and 0.465±0.048 mW cm−2, respectively), indicating that the catalytic structures with enzyme molecules improved EBC performance. Meanwhile, for evaluating the glucose effects, the polarization curves of EBCs using enzyme catalysts were measured at both with glucose and without glucose states. MPD of the EBC with glucose was 1.4-fold more than that of the EBC without glucose (MPDs of the former and latter were 0.465±0.048 and 0.654±0.025 mW cm−2, respectively), indicating that glucose played a key role in enhancing EBC performance. Although both the enzyme catalyst and glucose affected the EBC performance, the enzyme catalyst played a more dominant role than the glucose fuel, showing that utilization and optimization of enzyme catalysts was more effective for enhancing the EBC performance.
The stabilities of the CNT/PEI/[PCA/GOx], CNT/[PCA/GOx] and CNT/PEI/GOx catalysts were estimated by regularly measuring their catalytic activities (FAD redox reaction peaks) and their MPDs (with Pt/C as the cathode and Nafion 117 as the MEA) for 4 weeks (Figure 8). In terms of catalytic activity, the catalytic activities of the enzyme structures including PCA (CNT/PEI/PCA/GOx and CNT/PCA/GOx) were well maintained compared with those of CNT/PEI/GOx. After 4 weeks, the catalytic activities of CNT/PEI/PCA/GOx, CNT/PCA/GOx and CNT/PEI/GOx were 95%, 90% and 87% (anode) and 95%, 91% and 87% (cathode) of their initial values, respectively (Figures 8a and b). In addition, MPDs of the EBCs using the above catalysts were 93, 90 and 87% of their initial value (Figure 8c).

Stability measurements of CNT/PEI/GOx, CNT/[PCA/GOx] and CNT/PEI/[PCA/GOx] structures estimated using regular measurements of their (a) FAD oxidation reaction peaks, (b) FAD reduction reaction peaks and (c) maximum power densities for 4 weeks.
Table 1 shows comparison data between our CNT/PEI/[PCA/GOx] catalyst and other state-of-the-art GOx-based catalysts regarding their (i) biosensor and/or EBC performances and (ii) catalytic activities. From the comparison, the electrochemical performance and catalytic activity of the CNT/PEI/[PCA/GOx] were higher than other structures reported previously.
Taken together, it was concluded that using PCA, particularly in the form of PCA/GOx composite, was very useful for improving performance and stability of the EBC, as well as the catalytic activity of the enzyme catalysts due to concurrence of hydrophobic interactions, π-hydrogen bonds and π-conjugated electron transfer pathways that were newly caused by the PCA/GOx composite.
Conclusion
In this study, a novel enzyme catalyst using PCA (CNT/PEI/[PCA/GOx]) was developed, and its role as catalyst was verified by measuring its performance and stability within EBC. When PCA was included in the catalyst structure, the amount of immobilized GOx increased (3.3 U mg−1), and the electron transfer rate constant between PCA/GOx and CNT/PEI was enhanced (11.51 s−1). As a result, the EBC with CNT/PEI/[PCA/GOx] led to the best catalytic activity, EBC performance and catalytic stability due to effects of (i) electron collection via hydrophobic interactions, (ii) electron transfer pathways from π-conjugated bonds and (iii) enzyme stabilization due to π-hydrogen bonds. Regarding the hydrophobic interactions, physical entrapments between hydrophobic pockets near active sites of GOx and PCA were formed by hydrophobic interaction. Electrons produced in the active sites were captured without denaturation and transferred to the CNT/PEI supporter. Regarding the π-conjugated electron transfer pathways, interactions between the aldehyde groups of PCA and the amine groups of CNT/PEI supporter formed C=N bonds that were rigidly attached to the GOx, resulting in π-conjugated electron transfer pathways. Regarding the π-hydrogen bonds, the free pyrene groups of PCA attached on the surface of GOx interacted with the amine groups of PEI to form π-hydrogen bonds, which played a role in preventing denaturation of the PCA/GOx composite from CNT/PEI. The three attributes were analyzed using XPS and enzyme activity measurements. Based on the characterizations, the CNT/PEI/[PCA/GOx] structure yielded a high EBC MPD (2.1 mW cm−2) and long-lasting stability (maintaining 93% of the initial MPD after 4 weeks). We believe that our findings regarding three aspects of the new bonding mechanisms within the CNT/PEI/[PCA/GOx] structure will contribute to a protocol for establishing baseline processes of GOx-based biocatalysts.
References
Hoel, M. & Kvemdokk, S. Depletion of fossil fuels and the impacts of global warming. Resour. Energy Econ. 18, 115–136 (1996).
Minteer, S. D., Liaw, B. Y. & Cooney, M. J. Enzyme-based biofuel cells. Curr. Opin. Biotechnol. 18, 228–234 (2007).
Choi, H. S., Kim, D. S., Thapa, L. P., Lee, S. J., Kim, S. B., Cho, J., Park, C. & Kim, S. W. Production and characterization of cellobiose dehydrogenase from Phanerochaete chrysosporium KCCM 60256 and its application for an enzymatic fuel cell. Korean J. Chem. Eng. 34, 3434–3441 (2016).
Southcott, M., MacVittie, K., Halamek, J., Halamkova, L., Jemison, W. D., Lobeld, R. & Katz, E. A pacemaker powered by an implantable biofuel cell operating under conditions mimicking the human blood circulatory system–battery not included. Phys. Chem. Chem. Phys. 15, 6278–6283 (2013).
Rapoport, B. I., Kedzierski, J. T. & Sarpeshkar, R. A glucose fuel cell for implantable brain–machine interfaces. PLoS ONE 7, 1–15 (2012).
MacVittie, K., Halamek, J., Halamkov, L., Southcott, M., Jemison, W. D., Lobeld, R. & Katz, E. From ‘cyborg’ lobsters to a pacemaker powered by implantable biofuel cells. Energy Environ. Sci. 6, 81–86 (2013).
Rao, J. R. & Richter, G. Implantable bio-electrochemical power sources. Naturwissenschaften 61, 200–206 (1974).
Rao, J. R., Richter, G. J., Von Sturm, F. & Weidlich, E. The performance of glucose electrodes and the characteristics of different biofuel cell constructions. Bioelectrochem. Bioenerg. 3, 139–150 (1976).
Kwon, K. Y., Youn, J., Kim, J. H., Park, Y., Jeon, C., Kim, B. C., Kwon, Y., Zhao, X., Wang, P., Sang, B. I., Lee, J., Park, H. G., Chang, H. N., Hyeon, T., Ha, S., Jung, H. T. & Kim, J. Nanoscale enzyme reactors in mesoporous carbon for improved performance and lifetime of biosensors and biofuel cells. Biosens. Bioelectron. 26, 655–660 (2010).
Shimizu, K. & Ishihara, M. Immobilization of cellulolytic and hemicellulolytic enzymes on inorganic supports. Biotechnol. Bioeng. 29, 236–241 (1987).
Wei, Y., Xu, J., Feng, Q., Lin, M., Dong, H., Zhang, W. & Wang, C. J. A novel method for enzyme immobilization: direct encapsulation of acid phosphatase in nanoporous silica host materials. Nanosci. Nanotechnol. 1, 83–93 (2001).
Szymańska, K., Bryjak, J. & Jarzębski, A. B. Immobilization of invertase on mesoporous silicas to obtain hyper active biocatalysts. Top. Catal. 52, 1030–1036 (2009).
Schoevaart, R., Wolbers, M. W., Golubovic, M., Ottens, M., Kieboom, A. P. G., van Rantwijk, F., van der Wielen, L. A. M. & Sheldon, R. A. Preparation, optimization, and structures of cross‐linked enzyme aggregates (CLEAs). Biotechnol. Bioeng. 87, 754–762 (2004).
Lee, W. Y., Lee, K. S., Kim, T. H., Shin, M. C. & Park, J. K. Microfabricated conductometric urea biosensor based on sol‐gel immobilized urease. Electroanalysis 12, 78–82 (2000).
Tran, D. N. & Balkus, K. J. Perspective of recent progress in immobilization of enzymes. ACS Catal. 1, 956–968 (2011).
Balint, R., Cassidy, N. J. & Cartmell, S. H. Conductive polymers: towards a smart biomaterial for tissue engineering. Acta Biomater. 10, 2341–2353 (2014).
Decher, G. Fuzzy nanoassemblies: toward layered polymeric multicomposites. Science 277, 1232–1237 (1997).
Cash, K. J. & Clark, H. A. Nanosensors and nanomaterials for monitoring glucose in diabetes. Trends Mol. Med. 16, 584–593 (2010).
Le Goff, A., Gorgy, K., Holzinger, M., Haddad, R., Zimmerman, M. & Cosnier, S. Tris(bispyrene-bipyridine) iron(II): a supramolecular bridge for the biofunctionalization of carbon nanotubes via π-stacking and pyrene/β-cyclodextrin host–guest interactions. Chem. Eur. J. 17, 10216–10221 (2011).
Klein, D. R. Organic Chemistry. 2nd edn, (Wiley, New York, NY, 2013).
Yuan, C. X., Fan, Y. R., Zhang, T., Guo, H. X., Zhang, J. X., Wang, Y. L., Shan, D. L. & Lu, X. Q. A new electrochemical sensor of nitro aromatic compound based on three-dimensional porous Pt–Pd nanoparticles supported by graphene-multiwalled carbon nanotube composite. Biosens. Bioelectron. 58, 85–91 (2014).
Kuang, Y., Cui, Y., Zhang, Y., Yu, Y., Zhang, X. & Chen, J. A strategy for the high dispersion of PtRu nanoparticles onto carbon nanotubes and their electrocatalytic oxidation of methanol. Chem. Eur. J. 18, 1522–1527 (2012).
Sanderson, R. T. Chemical Bonds and Bond Energy, (Academic Press, New York, NY, London, 1971).
Hyun, K. H., Han, S. W., Koh, W. G. & Kwon, Y. Fabrication of biofuel cell containing enzyme catalyst immobilized by layer-by-layer method. J. Power Sources 286, 197–203 (2015).
Patil, R. D. & Adimurthy, S. Catalytic methods for imine synthesis. Asian J. Org. Chem. 2, 726–744 (2013).
Janegitz, B. C., Pauliukaite, R., Ghica, M. E., Brett, C. M. A. & Fatibello-Filho, O. Direct electron transfer of glucose oxidase at glassy carbon electrode modified with functionalized carbon nanotubes within a dihexadecylphosphate film. Sens. Actuator B 158, 411–417 (2011).
Hyun, K. H., Han, S. W., Koh, W. G. & Kwon, Y. Direct electrochemistry of glucose oxidase immobilized on carbon nanotube for improving glucose sensing. Int. J. Hydrogen Energy 40, 2199–2206 (2015).
Christwardana, M. & Kwon, Y. Effects of multiple polyaniline layers immobilized on carbon nanotube and glutaraldehyde on performance and stability of biofuel cell. J. Power Sources 299, 604–610 (2015).
Chung, Y., Hyun, K. & Kwon, Y. Fabrication of a biofuel cell improved by the π-conjugated electron pathway effect induced from a new enzyme catalyst employing terephthalaldehyde. Nanoscale 8, 1161–1168 (2016).
Chung, Y., Ahn, Y., Christwardana, M. & Kwon, Y. Development of a glucose oxidase-based biocatalyst adopting both physical entrapment and crosslinking, and its use in biofuel cells. Nanoscale 8, 9201–9210 (2016).
Wooten, M., Karra, S., Zhang, M. & Gorski, W. On the direct electron transfer, sensing, and enzyme activity in the glucose oxidase/carbon nanotubes system. Anal. Chem. 86, 752–757 (2014).
Ivnitski, D., Artyushkova, K., Rincn, R. A., Atanassov, P., Luckarift, H. R. & Johnson, G. R. Entrapment of enzymes and carbon nanotubes in biologically synthesized silica: glucose oxidase‐catalyzed direct electron transfer. Small 4, 357–364 (2008).
Cai, C. & Chen, J. Direct electron transfer of glucose oxidase promoted by carbon nanotubes. Anal. Biochem. 332, 75–83 (2004).
Liu, Y., Wang, M., Zhao, F., Xu, Z. & Dong, S. The direct electron transfer of glucose oxidase and glucose biosensor based on carbon nanotubes/chitosan matrix. Biosens. Bioelectron. 21, 984–988 (2005).
Milton, R. D., Wu, F., Lim, K., Abdellaoui, S., Hickey, D. P. & Minteer, S. D. Promiscuous glucose oxidase: electrical energy conversion of multiple polysaccharides spanning starch and dairy milk. ACS Catal. 5, 7218–7225 (2015).
Yu, C. M., Yen, M. J. & Chen, L. C. A bioanode based on MWCNT/protein-assisted co-immobilization of glucose oxidase and 2,5-dihydroxybenzaldehyde for glucose fuel cells. Biosens. Bioelectron. 25, 2515–2521 (2010).
Wang, G., Thai, N. M. & Yau, S. T. Preserved enzymatic activity of glucose oxidase immobilized on an unmodified electrode. Electrochem. Commun. 8, 987–992 (2006).
Dudzik, J., Chang, W. C., Kannan, A. M., Filipek, S., Viswanathan, S., Li, P., Renugopalakrishnan, V. & Audette, G. F. Cross-linked glucose oxidase clusters for biofuel cell anode catalysts. Biofabrication 5, 1–17 (2013).
Lim, J., Malati, P., Bonet, F. & Dunn, B. Nanostructured sol–gel electrodes for biofuel cells. J. Electrochem. Soc. 154, A140–A145 (2007).
Kuwahara, T., Homma, T., Kondo, M. & Shimomura, M. Fabrication of enzyme electrodes with a polythiophene derivative and application of them to a glucose fuel cell. Synth. Met. 159, 1859–1864 (2009).
Acknowledgements
This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry and Energy (MOTIE) of the Republic of Korea (No. 20164030201060) and the Korea National Research Foundation (KNRF) and the Ministry of Science, ICT and Future Planning (MSIP) (No. 2016M1A2A2937143) of the Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the NPG Asia Materials website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Christwardana, M., Chung, Y. & Kwon, Y. A new biocatalyst employing pyrenecarboxaldehyde as an anodic catalyst for enhancing the performance and stability of an enzymatic biofuel cell. NPG Asia Mater 9, e386 (2017). https://doi.org/10.1038/am.2017.75
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/am.2017.75
This article is cited by
-
Two-dimensional polymeric cobalt phthalocyanine synthesized by microwave irradiation and its use for continuous glucose monitoring
Korean Journal of Chemical Engineering (2023)
-
Effects of the gold nanoparticles including different thiol functional groups on the performances of glucose-oxidase-based glucose sensing devices
Korean Journal of Chemical Engineering (2018)
-
Highly sensitive glucose biosensor using new glucose oxidase based biocatalyst
Korean Journal of Chemical Engineering (2017)
