
Highlights
-
The innovation of this work mainly lies in the auto-regulation of reactive oxygen species (ROS) balance effective by combination of ROS antibacterial and ROS scavenging anti-inflammatory functions with a core–shell nanoplatform (CeO2@ZIF-8/Au), which can not only achieve high antibacterial efficiency, but also promote wound healing, and provide a new idea for the nano-catalytic system in the field of wound healing of bacterial infection.
Abstract
Reactive oxygen species (ROS) plays important roles in living organisms. While ROS is a double-edged sword, which can eliminate drug-resistant bacteria, but excessive levels can cause oxidative damage to cells. A core–shell nanozyme, CeO2@ZIF-8/Au, has been crafted, spontaneously activating both ROS generating and scavenging functions, achieving the multi-faceted functions of eliminating bacteria, reducing inflammation, and promoting wound healing. The Au Nanoparticles (NPs) on the shell exhibit high-efficiency peroxidase-like activity, producing ROS to kill bacteria. Meanwhile, the encapsulation of CeO2 core within ZIF-8 provides a seal for temporarily limiting the superoxide dismutase and catalase-like activities of CeO2 nanoparticles. Subsequently, as the ZIF-8 structure decomposes in the acidic microenvironment, the CeO2 core is gradually released, exerting its ROS scavenging activity to eliminate excess ROS produced by the Au NPs. These two functions automatically and continuously regulate the balance of ROS levels, ultimately achieving the function of killing bacteria, reducing inflammation, and promoting wound healing. Such innovative ROS spontaneous regulators hold immense potential for revolutionizing the field of antibacterial agents and therapies.

Similar content being viewed by others


1 Introduction
The complex and dynamic nature of reactive oxygen species (ROS) is a source of fascination for scientists and researchers alike. ROS, comprising hydrogen peroxide (H2O2), superoxide anion (O2−), singlet oxygen (1O2), and hydroxyl radical (·OH), plays crucial roles in living organisms [1,2,3]. But ROS is a double-edged sword, with its effects varying depending on its concentration and duration of action. At low levels, ROS can function as secondary messengers, aiding in cell signaling pathways, mitotic responses, cell proliferation/migration/differentiation, and resistance to pathogen invasion [4,5,6,7]. However, high levels of ROS can disrupt the body's regulatory mechanisms, leading to DNA mutations, cell damage, and the development of related diseases such as aging, cancer, cardiovascular disease, diabetes, ischemic/hypoxic injury, trauma infection, and neurological diseases [8,9,10,11,12]. Therefore, it is critical to modulate ROS levels and keep a balance between ROS generation and scavenging.
Nanozymes, a type of nanomaterial that exhibit high-efficiency catalytic activity similar to enzymes. Among these nanozymes, those that catalyze the in-situ generation of ROS (such as oxidases and peroxidases) have been widely used in anti-tumor and the development of new nano-antibacterial agents [13,14,15,16,17,18,19]. Within the tumor microenvironment, ROS can regulate the phenotype and function of tumor cells and various immune cells, affecting multiple processes of tumor immune response [20,21,22,23,24]. This can be applied for tumor-targeting drug delivery systems [25, 26]. Surface-oxidized arsenene nanosheets [27] was fabricated with effective·O2− and 1O2 generation and glutathione consumption for targeted ROS burst for cancer treatment. In addition, ROS can also destroy multiple vital substances (such as nucleic acids, proteins, and lipids) that are essential for bacterial cell normal physiological activities through oxidative action, and nanozymes can efficiently eliminate drug-resistant bacteria and delay the emergence of bacterial resistance [28,29,30,31,32,33,34,35,36,37]. We have synthesized MnFe2O4@MIL/Au-GOx and ZIF-8/Au-GOx nanozymes that achieve high-efficiency bacterial killing by using a synergistic strategy of cascade catalysis to produce ROS (·OH) and consume glutathione in the bacteria [38, 39]. In contrast, nanozymes with ROS clearance ability, including catalase (CAT), superoxide dismutase (SOD) and glutathione peroxidase (GPx), can mimic the intracellular antioxidant defense system to remove various toxic ROS, and show good therapeutic effects on acute kidney injury (AKI), acute liver injury (AILI), and wound healing and other related diseases [40,41,42,43,44,45,46].
Although tremendous progress has been made in generating or removing ROS, balancing ROS levels remains a huge challenge. Recent advances offer exciting possibilities for balancing ROS levels. For example, in photodynamic therapy, pyro pheophytin-bound Mn3O4 nanozymes can not only remove ROS in normal tissues but also produce ROS in the tumor microenvironment [47]. With the help of PDT or PTT, nanoceria and MoS2-CeO2 nanomaterials can also be used to enhance antibacterial and antioxidant activity [48, 49]. These advances provide new directions, however, the above process still requires the trigger of an external force (light) to activate the ROS adjustment switch.
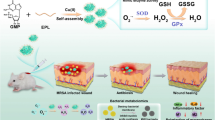
Herein, we have developed a core–shell nanozyme (CZA) that spontaneously active both ROS generating and scavenging functions, balancing ROS steady-state to promote wound healing. ZIF-8, as a metal–organic framework (MOF), has non-toxic and good biocompatibility, and mainly acts as a bridge between two different functional components. Firstly, The Au NPs on the shell of the ZIF-8 material rapidly exhibit its POD-like activity, utilizing the characteristics of high-efficiency of producing ROS to kill bacteria. Meanwhile, CeO2 core is encapsulated in ZIF-8, temporarily limiting its SOD and cat-like activity. Subsequently, as the ZIF-8 structure gradually decomposes in the acidic microenvironment, the CeO2 core is gradually released, exerting its SOD and CAT-like activity to eliminate excess ROS produced by the Au NPs. These two functions automatically and continuously regulate the balance of ROS levels, ultimately achieving the function of killing bacteria, reducing inflammation, and promoting wound healing (Fig. 1).

CeO2@ZIF-8/Au NPs treatment and healing promoting mechanism of bacterial infected wound in mice and equations of ROS production/clearance
2 Experimental Section
2.1 Materials
Cerium acetate (≥ 99%, (CH3COO)3Ce), 4′,6-diaminidine-2-phenylindole and propidium iodide bought in Jiangsu KeyGEN BioTECH Corp. 5,5-dimethyl-1-pyrrolin-N-oxide was bought in Shanghai Aladdin Biochemical Technology Co. Zinc nitrate hexahydrate (≥ 99.7%), chloroauric acid tetrahydrate (≥ 47.8%), sodium borohydride (96%), sodium chloride (99.7%) were purchased from Sinophinacea Chemical Reagents Co., LTD. Polyvinylpyrrolidone (PVP; Mw = 3500) was purchased from Aladdin. N,N-dimethylformamide (DMF), hydrogen peroxide (30%, H2O2), sodium hydroxide, hydrochloric acid and absolute ethanol (≥ 99.7%, C2H6O) were purchased from Xilong Technology Co., LTD. LB Broth, which contains peptone, yeast extract, and NaCl, was procured from Sangon Biotech. LB broth agar, which contains peptone, yeast extract, agar, and sodium chloride, was bought in Dalian Meilun Biotechnology Co. 2-methylimidazole was bought in Onokai Technology Co. Solutions are formulated via ultrapure H2O (18.2 MΩ cm).
2.2 Characterization
X-ray diffraction (XRD) were tested on Bruker SmartLab SE X-ray powder diffractometer. UV–vis spectra were tested on Shimadzu Cary 5000 UV–vis spectrophotometer. Transmission electron microscopy (TEM) photographs were acquired on JEM-1400, while elemental data obtained using Tecnai F30. Scanning electron microscopy (SEM) photographs were characterized using ZEISS Sigma SEM at 15 kV. Hydrodynamic diameter and ζ potential were determined via DLS on Malvern ZEN 3600. Reactive oxygen species was measured with ESR (EMX Bruker-10/12). Fluorescence imaging was recorded using a Leica DM 6000 B, and bacterial counts and area calculations were performed using NIH ImageJ software.
2.3 Preparation and Characterization of CeO2 Nanoparticles
CeO2 nanoparticles were synthesized using the solvothermal method. Initially, 0.1 M Ce(Ac)3 and 0.02 M NaOH were dissolved in 80 mL of ultrapure H2O, stirred until completely homogenized. The resultant mixture was then transferred to a high-pressure reaction vessel. The reaction was carried out at 180 °C for 24 h. The obtained product was purified 3 times using ultrapure H2O at 12,000 rpm and subsequently dried under vacuum to obtain CeO2 nanoparticles.
2.4 Preparation of CeO2@ZIF-8 Nanoparticles
CeO2 nanoparticles (35 mg) and PVP (1 g) were separately dissolved in methanol (50 mL), and then mixed for 24 h at 25 °C. The product was centrifuged and redispersed in 50 mL of 25.6 mM 2-methylimidazole (in methanol solvent) immediately, and then was stirred for 20 min at 25 °C until it was thoroughly mixed. Subsequently, we added Zn (NO3)2·6H2O (50 mL of 25.2 mM in methanol solvent) and mixed for an additional 30 min until complete reaction occurred to yield the product. The final product was centrifuged at 12,000 rpm, purified 3 times with methanol, and vacuum-dried.
2.5 Preparation of CeO2@ZIF-8/Au Nanoparticles
To synthesize CZA nanoparticles, we employed the reduction method. Firstly, 50 mg of prepared CZ NPs were dispersed evenly in methanol (50 mL) by sonication for 20 min. Next, added 150 μL of 0.1 M HAuCl4·3H2O and ultrasonic mixed for 20 min, then moved to a magnetic stirring device and stirred at 800 rpm. During the stirring process, quickly added freshly prepared NaBH4 solution (0.1 M, 3.75 mL), and the stirring speed of 800 rpm was maintained for another hour. The samples were centrifuged at 6000 rpm, purified 3 times with methanol, and vacuum-dried for 12 h to obtain the final product, CZA nanoparticles.
2.6 Hydroxyl Radical Assay and ESR Assay for Free Radical Scavenging
DMPO acts as a free radical catcher that can trap hydroxyl radicals and form adducts that can be detected by an ESR spectrometer. In the experiment, CZA (200 μg mL−1), DMPO (50 mM) and of H2O2 (5 mM) were thoroughly mixed. After 10 min of reaction, ESR signal was detected and recorded. ·OH in the free radical scavenging experiment was generated from FeCl2 (1.8 mM) and H2O2 (5 mM). The ESR signal was detected and recorded by adding 200 μg mL−1 of CZA in PBS buffer (pH = 4.5) for 4–24 h at 25 °C and reacting for 30 min.
2.7 Catalytic Analysis
In this experiment, the catalytic activity of the materials was evaluated using 3,3′,5,5′-tetramethylbenzidine (TMB) as an indicator. The colorimetric reaction was performed in a quartz cuvette using a mixture containing 500 μmol mL−1 H2O2, 3.2 mM TMB, NaAc buffer solution, and 200 μg mL−1 of the material (CeO2, CZ, and CZA). After the color stabilized, the UV absorption spectra of the solution were measured and recorded in the range of 350–800 nm.
TMB was also used as the indicator, and the steady-state kinetics of the material was analyzed by UV–vis spectrophotometer in a quartz cuvette. Firstly, the impacts of different pH values on the catalytic property of ZIF-8/Au were studied. Prepare 20 mM NaAc buffer, then adjust its pH with hydrochloric acid, and prepare NaAc buffer with pH = 3.5, 4.5, 5.5, 6.5 for use. Add 3.2 mM TMB, CZA and NaAc buffer to the UV dish, stir evenly, and place in the UV detection device. Finally add H2O2, stir quickly, and detect immediately at 652 nm in UV–vis spectrophotometry. The change curves of ultraviolet absorption intensity of the system were recorded within 0–10 min. Set the pH of NaAc buffer to 4.5 and the H2O2 concentration of 5, 12.5, 25, and 50 mM, respectively. We used Eqs. (1–3) as below:
Here A is absorbance value, and K represent the molar extinction coefficient, b is the thickness of the absorption layer, c is substrate’s concentration. Here, k = 39,000 M cm−1, and b = 1 cm.
2.8 In Vitro Antibacterial Assays
In the context of antimicrobial research, E. coli and S. aureus were selected as representative Gram-negative and Gram-positive bacteria, respectively. We procured Strains of E. coli (ATCC25922) and S. aureus (ATCC29213) in Xiang'an Hospital. Bacterial culture medium utilized LB broth of 25 g L−1 and pH of 4.5. In addition, the solid agar medium was constructed using ultrapure water prepared with agar powder involving LB broth (0.04 g mL−1). MIC and MBC against E. coli were determined with or without H2O2. Various concentrations of CZA (15, 30, 60, 120, 240 μg mL−1), 100 μmol L−1 of H2O2 and 106 CFU mL−1 of E. coli were blended in the above broth solution and maintained at 37 °C overnight. MIC against E. coli was determined by microplate reader at 595 nm. After diluting bacterial suspension 106-fold, which incubated on agar plates for sixteen hours, counted number of bacterial colonies, and established MBC against E. coli. Additionally, the material concentration for Staphylococcus aureus was adjusted to 1.25, 2.5, 5, 10, and 20 μg mL−1.
Then 106 CFU/mL of E. coli were treated with normal saline, H2O2, CeO2, CZ, CZA, CZA + H2O2 (the concentrations of CeO2, CZ, and CZA were 120 μg mL−1, respectively, and the concentration of H2O2 was 100 μmol L−1). The bacterial suspension was maintained at 37 °C for 16 h and subsequently diluted 106-fold. The diluted suspension was then cultured on agar plates for another 16 h to facilitate the enumeration of bacterial colonies. Similarly, Staphylococcus aureus was handled in the same manner, and the concentrations of CeO2, CZ, and CZA were adjusted to 10 μg mL−1.
2.9 SEM Morphology of Bacterial Specimens
Bacterial cells collected via centrifugation (5000 rpm, 5 min) and purified 2–3 times with PBS (pH = 7.4). Next, soaked in 2.5% glutaraldehyde and kept at 4 °C for 1 h. The fixed specimens were then progressively dehydrated with an increasing concentration of ethanol solution (10%, 30%, 50%, 70%, and 90%) for ten minutes. In the end, the specimens were dispersal in absolute ethanol.
2.9.1 TEM of Bacterial Specimens
Bacterial cells collected via centrifugation (5000 rpm, 5 min) and purified 2–3 times with PBS (pH = 7.4). Next, soaked in 2.5% glutaraldehyde and kept at 4° for 1 h. The bacterial specimens were then embedded in agar, sectioned with an ultrathin microtome and observed by TEM.
2.9.2 Live/Dead Bacteria Stain
Bacterial cells centrifuged and purified via sterile PBS. Subsequently, the treated bacteria were stained with 1.5 μmol L−1 of 4',6-diaminine-2-phenylindole (DAPI) and 1.6 μmol L−1 of propidium iodide (PI) for ten minutes. Purified bacteria via sterilized PBS to remove any free dye and surveyed through polarizing fluorescence microscope.
2.10 Protein Leakage
Purified bacterial cells with sterilized PBS. Collected the supernatant by centrifugation at 4 °C and added into 96-well plate. Then measured the protein concentration at 595 nm using the BCA Protein Detection Kit and Microplate Reader.
2.11 Cell Culture
L929 cells and 293T cells were grown in MEM (contain non-essential amino acid, PM150410, Pricella) or DMEM (SH30243.01, HyClone) medium containing 10% FBS (FBS500-A, HYCEZMBIO), penicillin streptomycin solution (1:100, SXQ006, HyClone), respectively. The cells were incubated in a humidified incubator at 37 °C with 5% CO2. Subcultures of cells were performed at 80–90% confluence using 0.25% trypsin–EDTA (SH30042.01, HyClone). CZA was immersed in PBS (pH 4–4.5) for 2 h (CZA-2 h) or 8 h (CZA-8 h), respectively, to degrade the ZIF-8 structure in the acidic microenvironment. 200 μM H2O2 and 2 μg mL−1 LPS (Sigma-Aldrich, L2880) was applied to damage the L929 cells. Materials CeO2, CZ, CZA, CZA-2 h, CZA-8 h with a concentration of 80 ug mL−1 were added to the cell culture medium.
2.12 Cytotoxicity Assay
L929 cells or 293T cells were carefully seeded in a 96-well plate at a density of 5 × 105 cells per well and allowed to grow for 24 h, respectively. 30 and 60 ug mL−1 CZA were incubated in cell culture medium. After 12, 24, or 48 h incubation, the cells viabilities were investigated through a Cell Counting Kit-8 assay (G4103-1ML, Servicebio). Cell apoptosis was further analyzed by Calcein-AM cell apoptosis assay (G1609-100T, Servicebio). After 50 μg mL−1 CZA were incubated in cell culture medium for 24 h, the cells in each group were suspend using 0.25% trypsin–EDTA. Then the cells were incubated in Calcein-AM and PI for 30 min and were observed with a fluorescence microscope at 490 and 545 nm laser to identify the live and dead cells. Take 4 views from each culture dish for observation, with a total of 12 views from each group. Count and count the number of dead and surviving cells in each field separately. ImageJ software is used to count cell numbers. After cells in each group were fixed with 4% paraformaldehyde (4 ℃, 15 min), phalloidin (G1041, Servicebio) was used to stain the cytoskeleton of cells, and DAPI (G1012, Servicebio) was used to stain the nucleus of cells. Confocal microscopic images are used to observe the cytoskeleton.
2.13 ROS Assays
After adding various materials to the cell culture medium for 24 h, the cell were washed by PBS buffer remove excess material. 200 μM H2O2 was applied to L929 cells for 1 h. After washing the cells again with PBS buffer, DCFH-DA fluorescent probe (S0033M, Boyetime) was utilized to detect the endogenous ROS levels of L929 cells. Finally, the cells were observed with a fluorescence microscope.
2.14 Real-Time PCR
Total RNA was extracted from L929 cells or 293T cells with ExTrizol Reagent (Protein Biotechnology, PR90) and reverse transcribed to cDNA by using PrimeScript™ RT reagent Kit with gDNA Eraser (RR047A, Takara). The qRT-PCR was performed on a LightCycler 480 RT-PCR system (Roche Diagnostics Ltd, Switzerland) with the TB Green® Premix Ex Taq™ (RR420A, Takara). The following primers were designed for each targeted mRNA or DNA:
L929 cell primers:
-
SOD1 (F): 5′- CACTCTAAGAAACATGGTGG-3′,-
-
SOD1 (R): 5′- GATCACACGATCTTCAATGG-3′,
-
SOD2 (F): 5′- CTTCAATAAGGAGCAAGGTC-3′,
-
SOD2 (R): 5′- CAGGTCTGACGTTTTTATACTG-3′,
-
GAPDH (F): 5′- GGGAAGCCCATCACCATCTTC-3′,
-
GAPDH (R): 5′- AGAGGGGCCATCCACAGTCT-3′,
-
Caspase-3 (F): 5′- AAGATACCGGTGGAGGCTGA -3′,
-
Caspase-3 (R): 5′-AAGGGACTGGATGAACCACG‐3′,
293T cell primers:
-
SOD1 (F): 5′-TGGTTTGCGTCGTAGTCTCC‐3′,
-
SOD1 (R): 5′-CTTCGTCGCCATAACTCGCT‐3′,
-
SOD2 (F): 5′-CAGGCAGCTGGCTCCGGTTT‐3′,
-
SOD2 (R): 5′-TGCAGTGGATCCTGATTTGG‐3′,
-
GAPDH (F): 5′- GCGAGATCCCTCCAAAATCAA-3′,
-
GAPDH (R): 5′- GTTCACACCCATGACGAACAT-3′,
-
Caspase-3 (F): 5′- GGAAGCGAATCAATGGACTCTGG-3′,
-
Caspase-3 (R): 5′- GCATCGACATCTGTACCAGACC-3′,
-
IL-6(F): ACTCACCTCTTCAGAACGAATTG,
-
IL-6(R): CCATCTTTGGAAGGTTCAGGTTG,
-
TNF-α(F): CCTCTCTCTAATCAGCCCTCTG,
-
TNF-α(R): GAGGACCTGGGAGTAGATGAG,
qPCR conditions consisted of an initial denaturing step of 5 min at 95 °C followed by 45 cycles of 10 s denaturation at 95 °C, 20 s annealing at 60 °C, and 20 s extension at 72 °C. The mRNA expression was normalized to the mRNA expression of GAPDH. The results were calculated using the comparative cycle threshold (ΔΔCt) method.
2.15 Cell Migration
The rate of cell migration plays an important role in wound healing, so in vitro scratch assays were performed to investigate the effect of synthetic materials on cell migration activity. A certain density of cell suspensions was seeded into 24-well plates, and the criterion was that cells could cover more than 90% of the well area after 24 h. After 24 h of culture, each well was scraped vertically with a 200 μL sterile pipette tip and washed three times with PBS to remove unattached cells. HUVECs cells were then cultured with DMEM containing CeO2, ZIF-8, CZ and CZA NPs with 5% FBS at a concentration of 80 μg mL−1 of the sample in each well. Cells cultured in the absence of samples were treated as controls. The purpose of adding 5% FBS to DMEM was to slow cell proliferation while maintaining cell survival to eliminate interference. After 24 h of incubation, the morphology of HUVECs cells was observed under an inverted fluorescence microscope. Migration assays were performed using imageJ software to quantitatively measure the area of the initial scratch (S0) and healing scratch (St). Mobility was calculated as Eq. (4):
2.16 Anti-infection Experiment In Vivo
All BALB/c mice used in the experiments were obtained from Beijing Vitarui Animal Technology Co., LTD and were 6–8 weeks old. All experiments complied with the rules in the Animal Central of Xiamen University. Mice were injected with 200 μL S. aureus containing 107 CFU mL−1. After 24 h of incubation of the bacteria subcutaneously, 4% chloral hydrate was used to anesthetize mice and a 7 mm diameter skin wound was cut at the injection site. Then those mice were stochasticly assigned into 7 groups. The above groups were set as one control group and six experimental groups. Among them, control set conducted with PBS (10 mM, 20 μL), meanwhile, 6 experimental groups were separately treated with 20 μL H2O2 (4 mM), 20 μL CeO2 (100 μg mL−1), 20 μL CZ (100 μg mL−1), 20 μL CeO2@ZIF-8/Au (100 μg mL−1), 10 μL H2O2, ZIF-8/Au (200 μg mL−1) + 8 μL H2O2 and 10 μL CeO2@ZIF-8/Au (200 μg mL−1) + 8 μL H2O2. The drug was given to cover the wounds of mice. The treatment day was set at day 0, and body weight and wound size were recorded every other day for all groups of mice. ImageJ software was used to measure the wound area. After 7 days, mice were sacrificed, and wounds, hearts, liver, spleen, lungs and kidneys were taken for H&E staining analysis.
3 Results and Discussion
3.1 Preparation and Characterization of CeO2@ZIF-8/Au Nanoparticles
Firstly, CeO2 nanoparticles were synthesized by solvothermal method. ZIF-8 was then allowed to grow on the outside of CeO2 nanoparticles to form CeO2@ZIF-8 (CZ). Finally, HAuCl4 was reduced by the original reduction method so that the Au nanoparticles were loaded on the surface of CZ. TEM revealed that the synthesized CeO2@ZIF-8/Au (CZA) nanoparticles presented rhomboid dodecahedral structure. The mean diameter is 150 ± 25 nm. CeO2 nanoparticles presented a cubic structure, and the average diameter is 10 ± 5 nm (Fig. 2a). From Fig. 2a, it can be seen that CeO2 nanoparticles are successfully wrapped in ZIF-8, and Au nanoparticles can be observed uniformly distributed on the surface of CZA after being loaded on CZ. At the same time, this process does not change the shape of nanoparticles. However, due to the acid etching of HAuCl4, the particle size of CZA is smaller than that of CZ. SEM images of CZA can also clearly observe its rhombohedral dodecahedron shape decorated with gold nanoparticles, which are consistent with TEM images. Furthermore, Fig. 2b, which shows the EDS data, illustrates the distribution of Ce, Zn, N, and Au throughout the entire CZA architecture. The presence of Ce can be attributed to CeO2, while Zn and N can be attributed to ZIF-8. Additionally, the presence of elemental Au is likely due to the gold nanoparticles.

Characterization of basic material properties. a TEM images of CeO2, CZ and CZA. SEM images of CZA. b High resolution image of CZA and EDS elemental mapping images (Ce, Zn, N, Au). c XRD of CeO2, ZIF-8, CZ and CZA. d Hydrodynamic diameter of CZA. e ζ-Potentials of CeO2, ZIF-8, CZ, and CZA (Dispersed in methanol, pH = 7.0). f FT-IR spectra of CeO2, ZIF-8, CZ and CZA
The X-ray diffraction (XRD) peaks of CZA is almost identical to that of CZ (Fig. 2c), and the position of the characteristic peak can exactly correspond to that of the standard card of CeO2 and the characteristic peak of ZIF-8, which suggests that the crystal structure of CZA remains unchanged compared to that of ZIF-8, and the crystal structure does not change during the loading of gold nanoparticles on the surface. However, in the XRD patterns of CZA, the characteristic peaks corresponding to the gold nanoparticles could not be found, which may be due to the fact that the gold nanoparticles are too small (~ 5 nm) to be detected. Dynamic light scattering analysis revealed that the mean size of hydrated particles in CZA was 250 ± 20 nm, as depicted in Fig. 2d. Through Zeta potential analysis, the ζ-Potentials of CeO2, ZIF-8, CZ, and CZA are + 43.67, + 15.20, − 23.97, and − 13.65 mV, respectively (Fig. 2e). Be able to tell that the formation of CZ changes the ζ-Potentials magnitude of CeO2 or ZIF-8. This phenomenon is similar to the literature [8], but the exact reason is unclear yet. Notably, the positively charged gold nanoparticle loading makes the ζ potential of CZA higher than that of CZ. Zeta potential analysis further confirmed the successful synthesis of CZA material. Figure 2f shows the Fourier transform infrared spectroscopy (FT-IR) spectra of each component of the material ranging from 4000 to 400 cm−1. The peaks appearing around 1407 and 1557 cm−1 are the stretching vibration peaks of Ce–O bond, and the peaks around 1645 cm−1 are the bending vibration peaks of H–O–H bond. The absorption peaks of 3134, 2925, and 1592 cm−1 in the infrared spectra of ZIF-8 are caused by the stretching vibration of aromatic C–H bond, the stretching vibration of fat C–H bond and the stretching vibration of C–N bond in 2-methylimidazole, respectively. The absorption peak at 420 cm−1 is the stretching vibration peak of the Zn–N bond, which is the bond formed by the coordination of Zn2+ and 2-methylimidazole. Among them, the absorption peak at 1592 cm−1 is slightly redshifted in the infrared spectra of CZ and CZA, and the characteristic absorption peak of CeO2 at 1407 cm−1 can also be observed in the infrared spectra of CZ and CZA. In short, the infrared spectra also indicate the successful synthesis of above materials.
3.2 Catalytic Performance of CeO2@ZIF-8/Au and ROS Scavenging
Figure 3a is a schematic diagram of the corresponding catalytic action of the inner (CeO2 NPs) and outer layers (Au NPs) of CZA. After the successful preparation of CZA, electron spin resonance spectroscopy was utilized to explore the ability of CZA to catalyze free radical production and free radical scavenging. To trap reactive oxygen species (ROS), 5,5-Dimethyl-1-Pyrroline N-oxide (DMPO) was used as a capture agent. The electron spin resonance test detected characteristic signals of the 1:2:2:1 ratio, confirming that the ROS generated by the CZA catalytic reaction are highly active and toxic hydroxyl radicals (·OH), as shown in Fig. 3b. This result ensures the effective bactericidal performance of CZA (Fig. 3b). The characteristic peak of ·OH produced by CZA catalysis shows some miscellaneous peaks, which may be due to the influence of organic components in ZIF-8 on the detection. In the fenton reaction group, the peak intensity of ·OH which produced by standard fenton reaction was high. However, after CZA (acid hydrolysis for 12 h at pH = 4.5 buffer) was added (Fenton + CZA group), the peak intensity of ·OH decreases significantly. This indicates that after acid immersion, the ZIF-8 framework of CZA is broken and inner CeO2 NPs were released, played the role of free radical scavenging.

Characterization of materials catalytic properties. a schematic diagram of the corresponding catalytic action of the inner (CeO2 NPs) and outer layers (Au NPs) of CZA. b ESR spectral of various groups. c UV–vis absorption of TMB in various groups (pH 4.5): TMB, H2O2 + TMB, CeO2 + H2O2 + TMB, CZA + TMB, CZ + H2O2 + TMB, CZA + H2O2 + TMB
Meanwhile, the catalytic performance of CZA was studied by UV–vis spectrophotometer with 3,3',5,5'-tetramethylbenzidine (TMB) as indicator. Figure 3c shows that only the CeO2 + H2O2 and CZA + H2O2 groups exhibited color reaction, with characteristic absorption peaks appearing in the UV–visible characteristic absorption peaks at 370 and 652 nm. CeO2 has suitable oxidation vacancies and the ability to rapidly convert Ce ions between tetravalent and trivalent ions. Thus, in acidic environments, CeO2 can exert peroxidase activity. POD activity of CeO2 catalyzes hydrogen peroxide to product · OH, which reacts with TMB to form oxTMB with two absorption peaks at 370 and 652 nm. Conversely, in Fig. 3c, the CZ + H2O2 group did not reveal characteristic absorption peaks at 370 and 652 nm, which indicated that CZ did not exhibit any simulated enzyme activity, further demonstrating the encapsulation of CeO2 in ZIF-8 frame structure can restrict the active site indicated by CeO2 and limit the catalytic performance of CZ. In addition, the absorbance intensity of CZA + H2O2 groups at 370 and 652 nm is the highest, indicate that the catalytic performance of Au nanoparticles is stronger than that of CeO2 nanoparticles.
Due to the significant impact of pH on the peroxidase-like activity of nanozymes, we investigated the influence of pH on the CZA catalytic performance, as illustrated in Fig. 4a. The data reveals that the catalytic performance of CZA gradually improves with decreasing pH, with significant enhancement observed when pH < 5. Therefore, in the subsequent experiments, the catalyzed reactions involving CZA were all carried out in the pH = 4.5 system.

Characterization of catalytic kinetics and acid degradation ability of materials a The absorbance value of TMB chromogram of CZA (100 μg mL−1) at varied pH values (3.5, 4.5, 5.5, and 6.5) when 5 mM H2O2 was added. Time-course absorbance of b Time-course absorbance of CZA with different H2O2 (50, 25, 12.5, and 5 mM) and Michaelis–Menten c kinetics and Lineweaver–Burk d curves of CZA. TEM images of CZA e, f after PBS treatment (pH = 7) for 4 and 8 h, g, h after acid treatment (pH = 4.5) for 4 and 8 h
Typical Mie steady-state kinetics was used to evaluate catalytic capacity of CZA. Figure 4b shows the change of UV absorbance intensity of CZA with time when the concentration of substrate H2O2 increases gradually. Based on the date in Fig. 4b, according to different substrate concentration and the calculated initial velocities (V0), the Michaelis–Menten equation was used to fit, and the fitting curve was drawn (Fig. 4c). Meanwhile, Fig. 4d was drawn using Mie equations based on Fig. 4c. As shown in Fig. 4c, d and based on the Mie equations, the Michaelis–Menten constant (Km) and the maximum velocity (Vmax) were 3.69 mmol L−1 and 1.73 × 10–8 M s–1.
In addition, we further studied the structural changes of CZA at different time in different pH environments by TEM. As is shown in Fig. 4e, f, after PBS treatment (pH = 7) for 4 and 8 h, no morphological changes occurred for CZA NPs. While after acid treatment (pH = 4.5) of CZA for 4 and 8 h, part of the structure of CZA gradually collapsed and released small nanoparticles with the average diameter of 10 ± 5 nm, which is corresponded with the size of CeO2 nanoparticles. This demonstrate that the ZIF-8 frame structure decomposed in the acidic microenvironment and released CeO2 nanoparticles from the core, which confirmed the design of automatically and continuously regulate the balance of ROS levels with CZA nanoplatform.
3.3 Evaluation of Antimicrobial Properties In Vitro
Due to the catalytic properties of CZA, we used Gram-negative strain E. coli and Gram-positive strain S. aureus to study the antibacterial performance. In a typical in vitro antimicrobial assay, both bacteria were cultured with a gradient increase in concentrations of CZA, while substrate H2O2 was provided at a concentration of 100 μmol L−1 in one of them to trigger the CZA catalytic reaction. After 16 h of incubation, tested the absorbance of the bacterial mixture at 595 nm using a microplate reader (OD595). The miscible liquids aforementioned were diluted at 106-fold and inoculated on solid LB agar plates to appraise bactericidal performance. The results depicted in Figs. 5a, b and S2 indicate a significant decrease in absorbance of both bacterial strains at OD595 with the increase in CZA concentration in the H2O2 group. The concentration in Fig. 5a is 120 μg mL−1, and the concentration in Fig. 5b is 10 μg mL−1. Finally, the OD595 curves of both bacteria in H2O2 group contacted with the OD595 curves of blank group (LB culture medium) at CZA concentrations of 120 and 10 μg mL−1, respectively. At the same time, the culture medium of the system showed a clear state at this concentration. The aforementioned concentration, 120 and 10 μg mL−1 for E. coli and S. aureus, respectively, represents the minimum inhibitory concentration (MIC) of CZA against these two bacterial strains. Minimum bactericidal concentration (MBC) were further colony counting on agar plates. The results (Fig. S2) showed that E. coli survival with 120 μg mL−1 CZA under H2O2 treatment was 0. Similarly, S. aureus survival with 10 μg mL−1 CZA was also 0, which is consistent with the findings shown in Fig. 5a, b. The above results indicate that, the MBC of CZA for E. coli was 120 and 10 μg mL−1 for S. aureus. In order to evaluate the antibacterial effects of CZA, we treated E. coli and S. aureus with PBS (control), H2O2, CeO2, CZ, CZA, and CZA + H2O2 at concentrations of 120 and 10 μg mL−1, respectively. Incubated overnight, the culture medium was diluted and cultured on agar plates. The results are presented in Fig. 5c. We also conducted statistical analysis of the bacterial colonies in each group and presented the results in Fig. 5d, e. The findings show that E. coli and S. aureus with CZA + H2O2 did not survive, while the bacterial growth in other groups was only weakly affected compared to the control group. Moreover, a cross-sectional index of bacterial membrane damage is protein leakage. Therefore, we detected the protein concentration using protein detection kits for bacteria treated with different groups (Fig. 5g). The protein leakage concentration was found to be the highest only in the bacteria treated with the CZA + H2O2 group (871 μg mL−1 for E. coli, 1029 μg mL−1 for S. aureus) and other groups did not detect protein leakage, indicating CZA can damage cell membranes in the presence of H2O2 and further confirming the antibacterial effect of CZA.

Characterization of antibacterial activity of materials in vitro. MIC of CZA NPs incubated with a E. coli and b S. aureus. c Agar plates photographs with remaining inoculated bacteria (above, E. coli; lower, S.aureus). d, e Bar chart of bacterial survival numbers. f TEM photographs of E. coli and S. aureus. g Protein exudation of bacterial in various group (mean ± SD, n = 3; P values: *P < 0.05, **P < 0.01, ***P < 0.001)
SEM (Fig. 6) and TEM (Fig. 5f) images were used to observe the morphology of bacteria treated by different groups. Observations reveal that in the bacteria treated with CZA + H2O2, the bacterial membranes appeared broken or sunken and were clearly visible. Additionally, the leaked cell contents caused some of the bacteria to stick to each other, resulting in incomplete bacterial structures. This phenomenon is attributed to the catalytic properties of CZA, which can convert the substrate H2O2 into toxic ·OH. Even at low dosages, the resulting ·OH is capable of directly damaging the bacterial membranes, divulgating cell contents. As such, CZA has a high potential for eradicating bacteria in various biological applications.

Live/Dead staining confocal microscopy and SEM photographs of E.coli and S.aureus after different treatments
To demonstrate the antibacterial properties of CZA more visually, we used DAPI and PI to stain all bacteria, and observed with polarized fluorescence microscope. DAPI could cross the cell walls and membranes of bacteria, and bind to the DNA inside the bacteria. PI cannot cross the cell membrane of bacteria, but can only enter the bacteria through the broken cell membrane, embedded in DNA, so it can only cause dead bacteria to emit red fluorescence. As can be seen from Fig. 6, for the two representative bacteria, only the CZA + H2O2 group showed strong red fluorescence, which means that in the presence of substrate H2O2, CZA could exert a broad spectrum of bactericidal activity. Meanwhile, the other groups only showed strong blue fluorescence, indicating that the bacteria treated by the other treatment groups did not have too much survival pressure.
3.4 In Vitro Cytotoxicity
To further investigate CZA cytotoxicity in vitro, we treated 293T cells and L929 cells at concentrations of 30, 60, and 120 μg mL−1 for 12, 24, and 48 h, respectively. Using the CCK-8 kit, it was observed that there was no significant decrease in cell viability of both L929 and 293T cell lines at 60 or 120 μg mL−1 even after 48 h of treatment (Fig. 7a, b), which suggested that CZA may not have significant cytotoxicity on cells. To further investigate whether CZA incubation leads to cell death, Calcein-AM and PI staining were performed on 293T cells and L929 cells incubated with CZA for 24 h (Fig. 7c–f). Cells that undergo necrosis or are in the final stage of apoptosis will be stained red by PI. Compared with the control group, there was no statistically significant difference in the cell survival rate of L929 cells or 293T cells incubated with CZA for 24 h. To observe whether CZA affects cytoskeleton of cells or not, the cytoskeleton of L929 cells or 293T cells incubated with CZA for 48 h was stained with phalloidin. No significant toxic effects of CZA treatment on the cytoskeleton of L929 cells or 293T cells were observed using laser confocal microscopy (Fig. 7g). The above results suggest that CZA may have acceptable biological safety.

Material cytotoxicity data and live and dead cytoskeleton staining In vitro cytotoxicity and apoptosis investigation. a In vitro cytotoxicity assays for different concentrations and incubation times of CZA in L929 cells. b In vitro cytotoxicity assays for different concentrations and incubation times of CZA in 293T cells. c The apoptosis of L929 cells after CZA treatments was detected by calcein AM/PI staining (Green: live cells, Red: dead cells). Scale bars: 50 μm. d Statistical chart of L929 cell survival quantity. e The apoptosis of 293T cells after CZA treatments was detected by calcein AM/PI staining (Green: live cells, Red: dead cells). Scale bars: 50 μm. f Statistical chart of 293T cell survival quantity. g Cytoskeleton staining of L929 and 293T cells (Red: cytoskeleton, Blue: nucleus). Scale bars: 5 μm
3.5 Evaluation of Antioxidant Ability In Vitro
To evaluate whether CZA significantly enhances cellular antioxidant or inflammatory capacity in vitro, we treated cells with H2O2 or LPS for 1 h after CZA pre-incubated for 24 h, and then measured cell viabilities (Fig. 8a, b). Compared with the non-pre-incubated group (just treated by H2O2 or LPS), the increased cell viabilities were observed in pre-incubated CZA + LPS L929 cells (41.42%), pre-incubated CZA + H2O2 L929 cells (55.61%), pre-incubated CZA + LPS 293T cells (27.28%) and pre-incubated CZA + H2O2 293T cells (58.74%), which suggests that pre-incubation of CZA enhances the antioxidant or inflammatory abilities of L929 cells and 293T cells. We use ROS probe DCFH-DA to detect the production of reactive oxygen species in cells (pre-incubated with CeO2, ZIF8, CZA, CZA-2H, CZA-8H) treated with hydrogen peroxide. Fluorescence microscopy was used to observe of intracellular ROS probe luminescence in different treatment groups. Strong green fluorescence was observed in both group treated with H2O2 and ROS reagent that can cause strong oxidative stress in the kit (positive control), while in other group pre-incubated with CeO2, ZIF-8 or CZA, intracellular ROS levels decreased compared to the H2O2 group (Fig. 8c). In order to acidify CZA and release CeO2 to exert the antioxidant capacity of CZA, CZA soaked in PBS at pH 4–4.5 for 2 and 8 h, respectively. L929 cells were treated with H2O2 for 1 h after CZA (no soaking), CZA-2H, and CZA-8H pre-incubated 24 h. It was observed that CZA-8H can better reduce the production of ROS in L929 cells (Fig. 8c). This indicates that acid immersion can decomposition ZIF-8 framework and release encapsulated CeO2, leading to effectively remove ROS. In addition, if the material can promote cell migration and proliferation during the proliferative phase of wound healing, it will be very beneficial to promote wound healing. We further carried out the cell scratch experiment to investigate the ability of the material to promote cell migration after the effect of ROS scavenging. As shown in Fig. S4. compared with the control group, CZA NPs had certain promotion effect on cell migration. For a more intuitive demonstration, the healing rate of the scratched area was quantitatively calculated using the Image J software (Fig. S5). The data showed that the scratch mobility of CZA cultivated in cells for 24 h could reach 53%, which was almost reached the value of CeO2 NPs group. Cell scratch assay showed that CZA materials containing CeO2 NPs components can effectively promote cell migration, which is essential for wound healing. To understand how CZA helps cells reduce intracellular oxidative stress or resist inflammation, we further extracted mRNA from cells and evaluated transcriptional expression of antioxidant and apoptotic genes. Similar to previous experiments, cells were pre-incubated with CZA for 24 h or without pre-incubation, and then treated by LPS or H2O2. Compared with the control group, the upregulation of antioxidant related mRNA (SOD1 and SOD2) in the H2O2 treated L929 and 293T group showed activation of intracellular antioxidant mechanisms. However, in the CZA + H2O2 L929 and 293T group, the corresponding cellular expression levels of these mRNA were significantly down-regulated, which suggests that CZA helps cells remove ROS (Fig. 8d–i). After pre-incubation of CZA 24 h, the mRNA expression level of caspase-3 in L929 or 293T cells treated with H2O2 or LPS is lower than that in L929 or 293T cells not pre-incubated (Fig. 8f–k), which suggested that pre-incubation of CZA helps cells enhanced the anti-apoptotic ability. In addition, in order to better understand the performance of CZA in clearing ROS at the cellular level, we further extracted mRNA from cells and detected the expression levels of several inflammatory factors, including IL-6 and TNF-α. As shown in Fig. S6a, b. the levels of IL-6 and TNF-α in 293T cells were significantly increased after LPS was added, while the levels of inflammatory factors in cells were significantly decreased after CZA NPs treatment, indicating that CZA NPs can effectively alleviate inflammation.

Anti-inflammatory data characterization of materials In vitro CZA enhancing cell antioxidant function investigation. a Cells viabilities of L929 cells after the treatment with PBS, CZA, LPS, CZA + LPS, H2O2, and CZA + H2O2. b Cells viabilities of 293T cells after the treatment with PBS, CZA, LPS, CZA + LPS, H2O2, and CZA + H2O2. c The images of H2O2 induced L929 cells with oxidized DCFH-DA fluorescence treated with different components. Scale bars: 50 μm. d-f SOD1, SOD2, Caspase-3 gene expression level of L929 cell after treated with H2O2 or CZA + H2O2. g Caspase-3 gene expression level of L929 cell after treated with LPS or CZA + LPS. h–j SOD1, SOD2, Caspase-3 gene expression level of 293T cell after treated with H2O2 or CZA + H2O2. k Caspase-3 gene expression level of 293T cell treated with LPS or CZA + LPS. (mean ± SD, n = 3; P values: *P < 0.05, **P < 0.01, ***P < 0.001)
3.6 Wound Infection Treatment
In order to deeply explore the anti-microbial effects of CZA NPs on wound healing, we subcutaneously injected BALB/c mice with 200 μL of 107 CFU mL−1 S. aureus, as illustrated in Fig. 9a. Following a 24-h incubation period, a skin wound approximately 7 mm in diameter was formed at the injection site where the bacteria had been subcutaneously incubated. Stochasticly allotted infected mice into 7 sets of 3 per set, then handled with PBS, H2O2, CeO2, CZ, CZA, ZIF-8/Au (ZA) + H2O2, and CZA + H2O2, respectively. Using PBS treatment set as the control. Figure 9b, c presents digital photographs and the relative wound area with different treatments at various time periods (0, 2, 4, 6, and 7 days), with all mice exhibiting clear festering of the wounds on the first day. The mice exhibited a declining trend in weight during the first 1–3 days of infection (Fig. 9d), indicating that the bacterial infection to be worked. After treatment, the weight of mice began to show a steady upward trend all the time, indicating that after treatment, the material itself did not cause much harm to mice and had good biocompatibility. At the same time, the wound area of mice also showed a steady downward trend, especially in the first 3 days. Finally, on the seventh day, the wound area of mice treated with PBS, H2O2, CeO2, CZ, CZA, ZA + H2O2, CZA + H2O2 was reduced to 23.8%, 22.8%, 23.1%, 25.3%, 23.6%,15.6%, and 7.5% (Fig. 9c), respectively. Among them, Compared with ZA + H2O2 group, CZA + H2O2 group showed the better therapeutic effect. The above results indicate that the catalytic capacity of CZA plays a vital role in antibacterial therapy and demonstrated that the ROS scavenging function of CeO2 NPs played an additional promoting effect on wound healing after the completion of ROS antimicrobial treatment.

CZA antibacterial/promote the healing of animal models. a Schematic diagram of anti-infection implementation scenario in vivo for CZA. b Wound area graph treated differently along time points. Scale bars: 5 mm. c Corresponding changes in relative wound area. d Mice body weight. e H&E stained photographs at wound site (gules and yellow arrows mean hair follicles and blood vessels) (mean ± standard deviation, n = 3; P values: *P < 0.05, **P < 0.01, ***P < 0.001)
In order to obtain more reliable details and further appraise the wound healing process, we took skin tissues from the wound and subjected to H&E staining, as shown in Fig. 9e. Consistent with the digital photographs, glucose-treated mice exhibited partial skin tissue regeneration and some subcutaneous hemorrhage, stating that the wound was not fully closed. The tissue regeneration of mice in PBS, ZIF-8, CZ, CZA, and ZA was similar and the tissue structure was good. Abundantly distributed capillaries provide nutrients, indicating rapid regeneration of skin tissue. In contrast, CZA and H2O2-treated mice showed good skin structure and reduced capillaries, indicating utter regeneration of the wound site. In addition, the histopathological photographs in Fig. 10 revealed that the major organs of infected or treated mice were not conspicuously damaged. In summary, based on the appraisement of wound bactericidal properties, CZA NPs exhibited the ability of bacterial removal and wound healing by effectively killing bacteria and accelerating the healing process of infected wounds in the presence of H2O2. In addition, CZA NPs exhibit good biocompatibility, making them promising candidates for the treatment of infected wounds.

Images of H&E-stained heart, liver, spleen, lung, and kidney obtained from mice of PBS, CZA, and CZA + H2O2 groups
4 Conclusions
We designed a dual synergetic MOF nanozyme (CeO2@ZIF-8/Au), which possesses both antibacterial and ROS regulating functions for wound repair. Firstly, it can catalyze and product ·OH to complete the efficient antibacterial effect to kill bacteria. Then, as the ZIF-8 structure decomposes in the acidic microenvironment, the CeO2 core is gradually released, which can effectively remove ROS and auto-regulate ROS balance, realize the purpose of reducing inflammation and promote wound healing. The innovation of this work mainly lies in the auto-regulation of ROS balance effective by combination of ROS antibacterial and ROS scavenging anti-inflammatory functions with CeO2@ZIF-8/Au nanoplatform, which can not only achieve high antibacterial efficiency, but also promote wound healing, and provide a new idea for the nano-catalytic system in the field of wound healing of bacterial infection. Besides, such innovative ROS spontaneous regulators hold immense potential for revolutionizing the field of cancer therapies [49,50,51,52,53,54,55,56,57,58], anti-SARS-CoV-2 and other viruses [59,60,61], and much broader biomedical applications due to the fundamental importance of ROS in health regulation.
References
H. Sies, V.V. Belousov, N.S. Chandel, M.J. Davies, D.P. Jones et al., Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 23, 499–515 (2022). https://doi.org/10.1038/s41580-022-00456-z
P. Strzyz, ATP and ROS signal cell extrusion. Nat. Rev. Mol. Cell Biol. 23, 387 (2022). https://doi.org/10.1038/s41580-022-00487-6
Z. Liu, T.S. Xiao, Partners with a killer: metabolic signaling promotes inflammatory cell death. Cell 184, 4374–4376 (2021). https://doi.org/10.1016/j.cell.2021.07.036
C. Lennicke, H.M. Cochemé, Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 81, 3691–3707 (2021). https://doi.org/10.1016/j.molcel.2021.08.018
S.G. Rhee, H2O2, a necessary evil for cell signaling. Science 312, 1882–1883 (2006). https://doi.org/10.1126/science.1130481
C. Tapeinos, A. Pandit, Physical, chemical, and biological structures based on ros-sensitive moieties that are able to respond to oxidative microenvironments. Adv. Mater. 28, 5553–5585 (2016). https://doi.org/10.1002/adma.201505376
H. Kawagishi, T. Finkel, Unraveling the truth about antioxidants: ROS and disease: finding the right balance. Nat. Med. 20, 711–713 (2014). https://doi.org/10.1038/nm.3625
S. Koo, M.G. Lee, A. Sharma, M. Li, X. Zhang et al., Harnessing GLUT1-targeted pro-oxidant ascorbate for synergistic phototherapeutics. Angew. Chem. Int. Ed. 61, e202110832 (2022). https://doi.org/10.1002/anie.202110832
D. Wei, Y. Yu, Y. Huang, Y. Jiang, Y. Zhao et al., A near-infrared-II polymer with tandem fluorophores demonstrates superior biodegradability for simultaneous drug tracking and treatment efficacy feedback. ACS Nano 15, 5428–5438 (2021). https://doi.org/10.1021/acsnano.1c00076
B. Steinhorn, A. Sorrentino, S. Badole, Y. Bogdanova, V. Belousov et al., Chemogenetic generation of hydrogen peroxide in the heart induces severe cardiac dysfunction. Nat. Commun. 9, 4044 (2018). https://doi.org/10.1038/s41467-018-06533-2
D.E. McCoy, T. Feo, T.A. Harvey, R.O. Prum, Structural absorption by barbule microstructures of super black bird of paradise feathers. Nat. Commun. 9, 1 (2018). https://doi.org/10.1038/s41467-017-02088-w
W.Y. Kim, M. Won, S. Koo, X. Zhang, J.S. Kim, Mitochondrial H2Sn-mediated anti-inflammatory theranostics. Nano-Micro Lett. 13, 168 (2021). https://doi.org/10.1007/s40820-021-00689-1
R. Li, T. Chen, X. Pan, Metal–organic-framework-based materials for antimicrobial applications. ACS Nano 15, 3808–3848 (2021). https://doi.org/10.1021/acsnano.0c09617
Y. Huang, J. Ren, X. Qu, Nanozymes: classification, catalytic mechanisms, activity regulation, and applications. Chem. Rev. 119, 4357–4412 (2019). https://doi.org/10.1021/acs.chemrev.8b00672
J. Li, S. Song, J. Meng, L. Tan, X. Liu et al., 2D MOF periodontitis photodynamic ion therapy. J. Am. Chem. Soc. 143, 15427–15439 (2021). https://doi.org/10.1021/jacs.1c07875
X. Ji, L. Ge, C. Liu, Z. Tang, Y. Xiao et al., Capturing functional two-dimensional nanosheets from sandwich-structure vermiculite for cancer theranostics. Nat. Commun. 12, 1124 (2021). https://doi.org/10.1038/s41467-021-21436-5
Z. Yang, D. Gao, J. Zhao, G. Yang, M. Guo et al., Thermal immuno-nanomedicine in cancer. Nat. Rev. Clin. Oncol. 20, 116–134 (2023). https://doi.org/10.1038/s41571-022-00717-y
M. Zandieh, J. Liu, Nanozymes: definition, activity, and mechanisms. Adv. Mater. (2023). https://doi.org/10.1002/adma.202211041
X. Liu, Q. Liu, X. He, G. Yang, X. Chen et al., NIR-II-enhanced single-atom-nanozyme for sustainable accelerating bacteria-infected wound healing. Appl. Surf. Sci. 612, 155866 (2023). https://doi.org/10.1016/j.apsusc.2022.155866
E.C. Cheung, K.H. Vousden, The role of ROS in tumour development and progression. Nat. Rev. Cancer 22, 280–297 (2022). https://doi.org/10.1038/s41568-021-00435-0
S. Wang, R. Tian, X. Zhang, G. Cheng, P. Yu et al., Beyond photo: xdynamic therapies in fighting cancer. Adv. Mater. 33, e2007488 (2021). https://doi.org/10.1002/adma.202007488
X.-L. Xu, N.-N. Zhang, G.-F. Shu, D. Liu, J. Qi et al., A luminol-based self-illuminating nanocage as a reactive oxygen species amplifier to enhance deep tumor penetration and synergistic therapy. ACS Nano 15, 19394–19408 (2021). https://doi.org/10.1021/acsnano.1c05891
Z. Chen, Y. Sun, J. Wang, X. Zhou, X. Kong et al., Dual-responsive triple-synergistic Fe-MOF for tumor theranostics. ACS Nano 17, 9003–9013 (2023). https://doi.org/10.1021/acsnano.2c10310
X. Zan, D. Yang, Y. Xiao, Y. Zhu, H. Chen et al., Facile general injectable gelatin/metal/tea polyphenol double nanonetworks remodel wound microenvironment and accelerate healing. Adv. Sci. (2023). https://doi.org/10.1002/advs.202305405
L. Liu, M. Bi, Y. Wang, J. Liu, X. Jiang et al., Artificial intelligence-powered microfluidics for nanomedicine and materials synthesis. Nanoscale 13, 19352–19366 (2021). https://doi.org/10.1039/d1nr06195j
M. Cao, N. Diao, X. Cai, X. Chen, Y. Xiao et al., Plant exosome nanovesicles (PENs): green delivery platforms. Mater. Horiz. 10, 3879–3894 (2023). https://doi.org/10.1039/d3mh01030a
N. Kong, H. Zhang, C. Feng, C. Liu, Y. Xiao et al., Arsenene-mediated multiple independently targeted reactive oxygen species burst for cancer therapy. Nat. Commun. 12, 4777 (2021). https://doi.org/10.1038/s41467-021-24961-5
D. Gao, T. Chen, S. Chen, X. Ren, Y. Han et al., Targeting hypoxic tumors with hybrid nanobullets for oxygen-independent synergistic photothermal and thermodynamic therapy. Nano-Micro Lett. 13, 99 (2021). https://doi.org/10.1007/s40820-021-00616-4
Y. Zhu, P. Xu, X. Zhang, D. Wu, Emerging porous organic polymers for biomedical applications. Chem. Soc. Rev. 51, 1377–1414 (2022). https://doi.org/10.1039/d1cs00871d
D. Zhang, D. Zhong, J. Ouyang, J. He, Y. Qi et al., Microalgae-based oral microcarriers for gut microbiota homeostasis and intestinal protection in cancer radiotherapy. Nat. Commun. 13, 1413 (2022). https://doi.org/10.1038/s41467-022-28744-4
J. Yang, X. Zhang, C. Liu, Z. Wang, L. Deng et al., Biologically modified nanoparticles as theranostic bionanomaterials. Prog. Mater. Sci. 118, 100768 (2021). https://doi.org/10.1016/j.pmatsci.2020.100768
P. Jangili, N. Kong, J.H. Kim, J. Zhou, H. Liu et al., DNA-damage-response-targeting mitochondria-activated multifunctional prodrug strategy for self-defensive tumor therapy. Angew. Chem. Int. Ed. 61, e202117075 (2022). https://doi.org/10.1002/anie.202117075
Z. Yang, D. Gao, X. Guo, L. Jin, J. Zheng et al., Fighting immune cold and reprogramming immunosuppressive tumor microenvironment with red blood cell membrane-camouflaged nanobullets. ACS Nano 14, 17442–17457 (2020). https://doi.org/10.1021/acsnano.0c07721
M. Deng, M. Zhang, R. Huang, H. Li, W. Lv et al., Diabetes immunity-modulated multifunctional hydrogel with cascade enzyme catalytic activity for bacterial wound treatment. Biomaterials 289, 121790 (2022). https://doi.org/10.1016/j.biomaterials.2022.121790
J. Ouyang, C. Feng, X. Zhang, N. Kong, W. Tao, Black phosphorus in biological applications: evolutionary journey from monoelemental materials to composite materials. Acc. Mater. Res. 2, 489–500 (2021). https://doi.org/10.1021/accountsmr.1c00039
W. Zhu, J. Mei, X. Zhang, J. Zhou, D. Xu et al., Photothermal nanozyme-based microneedle patch against refractory bacterial biofilm infection via iron-actuated Janus ion therapy. Adv. Mater. 34, e2207961 (2022). https://doi.org/10.1002/adma.202207961
G.W.K. Moore, S.E.L. Howell, M. Brady, X. Xu, K. McNeil, Anomalous collapses of Nares Strait ice Arches leads to enhanced export of Arctic Sea ice. Nat. Commun. 12, 1 (2021). https://doi.org/10.1038/s41467-020-20314-w
M. Wang, X. Zhou, Y. Li, Y. Dong, J. Meng et al., Triple-synergistic MOF-nanozyme for efficient antibacterial treatment. Bioact. Mater. 17, 289–299 (2022). https://doi.org/10.1016/j.bioactmat.2022.01.036
X. Zhou, S. Zhang, Y. Liu, J. Meng, M. Wang et al., Antibacterial cascade catalytic glutathione-depleting MOF nanoreactors. ACS Appl. Mater. Interfaces 14, 11104–11115 (2022). https://doi.org/10.1021/acsami.1c24231
Y. Sun, S. Mu, Z. Xing, J. Guo, Z. Wu et al., Catalase-mimetic artificial biocatalysts with Ru catalytic centers for ROS elimination and stem-cell protection. Adv. Mater. 34, e2206208 (2022). https://doi.org/10.1002/adma.202206208
Y. Ma, Z. Lu, B. Jia, Y. Shi, J. Dong et al., DNA origami as a nanomedicine for targeted rheumatoid arthritis therapy through reactive oxygen species and nitric oxide scavenging. ACS Nano 16, 12520–12531 (2022). https://doi.org/10.1021/acsnano.2c03991
Y. Zhao, S. Song, D. Wang, H. Liu, J. Zhang et al., Nanozyme-reinforced hydrogel as a H2O2-driven oxygenerator for enhancing prosthetic interface osseointegration in rheumatoid arthritis therapy. Nat. Commun. 13, 6758 (2022). https://doi.org/10.1038/s41467-022-34481-5
R. Hou, T. Lu, W. Gao, J. Shen, Z. Yu et al., Prussian blue nanozyme promotes the survival rate of skin flaps by maintaining a normal microenvironment. ACS Nano 16, 9559–9571 (2022). https://doi.org/10.1021/acsnano.2c02832
Z. Li, Y. Zhao, H. Huang, C. Zhang, H. Liu et al., A nanozyme-immobilized hydrogel with endogenous ROS-scavenging and oxygen generation abilities for significantly promoting oxidative diabetic wound healing. Adv. Healthc. Mater. 11, e2201524 (2022). https://doi.org/10.1002/adhm.202201524
D.Y. Zhang, M.R. Younis, H. Liu, S. Lei, Y. Wan et al., Multi-enzyme mimetic ultrasmall iridium nanozymes as reactive oxygen/nitrogen species scavengers for acute kidney injury management. Biomaterials 271, 120706 (2021). https://doi.org/10.1016/j.biomaterials.2021.120706
C. Zhou, L. Zhang, Z. Xu, T. Sun, M. Gong et al., Self-propelled ultrasmall AuNPs-tannic acid hybrid nanozyme with ROS-scavenging and anti-inflammatory activity for drug-induced liver injury alleviation. Small 19, e2206408 (2023). https://doi.org/10.1002/smll.202206408
M. Li, L. Huo, J. Zeng, G. Zhu, X. Liu et al., Switchable ROS scavenger/generator for MRI-guided anti-inflammation and anti-tumor therapy with enhanced therapeutic efficacy and reduced side effects. Adv. Healthc. Mater. 12, e2202043 (2023). https://doi.org/10.1002/adhm.202202043
Y. Sun, X. Sun, X. Li, W. Li, C. Li et al., A versatile nanocomposite based on nanoceria for antibacterial enhancement and protection from aPDT-aggravated inflammation via modulation of macrophage polarization. Biomaterials 268, 120614 (2021). https://doi.org/10.1016/j.biomaterials.2020.120614
T. Ma, X. Zhai, Y. Huang, M. Zhang, X. Zhao et al., A smart nanoplatform with photothermal antibacterial capability and antioxidant activity for chronic wound healing. Adv. Healthc. Mater. 10, e2100033 (2021). https://doi.org/10.1002/adhm.202100033
C. Feng, Z. Xiong, X. Sun, H. Zhou, T. Wang et al., Beyond antioxidation: harnessing the CeO2 nanoparticles as a renoprotective contrast agent for in vivo spectral CT angiography. Biomaterials 299, 122164 (2023). https://doi.org/10.1016/j.biomaterials.2023.122164
T. Hu, Z. Gu, G.R. Williams, M. Strimaite, J. Zha et al., Layered double hydroxide-based nanomaterials for biomedical applications. Chem. Soc. Rev. 51, 6126–6176 (2022). https://doi.org/10.1039/d2cs00236a
G. Parekh, Y. Shi, J. Zheng, X. Zhang, S. Leporatti, Nano-carriers for targeted delivery and biomedical imaging enhancement. Ther. Deliv. 9, 451–468 (2018). https://doi.org/10.4155/tde-2018-0013
C. Zhang, Y. Yu, S. Shi, M. Liang, D. Yang et al., Machine learning guided discovery of superoxide dismutase nanozymes for androgenetic alopecia. Nano Lett. 22, 8592–8600 (2022). https://doi.org/10.1021/acs.nanolett.2c03119
X. Zhang, Tea and cancer prevention. J. Can. Res. Updates 4, 65–73 (2015). https://doi.org/10.6000/1929-2279.2015.04.02.4
G. Huang, J. Zang, L. He, H. Zhu, J. Huang et al., Bioactive nanoenzyme reverses oxidative damage and endoplasmic reticulum stress in neurons under ischemic stroke. ACS Nano 16, 431–452 (2022). https://doi.org/10.1021/acsnano.1c07205
X. Zhang, G. Parekh, B. Guo, X. Huang, Y. Dong et al., Polyphenol and self-assembly: metal polyphenol nanonetwork for drug delivery and pharmaceutical applications. Future Drug Discov. 1, 0001 (2019). https://doi.org/10.4155/fdd-2019-0001
L. He, G. Huang, H. Liu, C. Sang, X. Liu et al., Highly bioactive zeolitic imidazolate framework-8-capped nanotherapeutics for efficient reversal of reperfusion-induced injury in ischemic stroke. Sci. Adv. 6, eaay9751 (2020). https://doi.org/10.1126/sciadv.aay9751
H.J. Forman, H. Zhang, Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 20, 689–709 (2021). https://doi.org/10.1038/s41573-021-00233-1
Z. Tang, X. Zhang, Y. Shu, M. Guo, H. Zhang et al., Insights from nanotechnology in COVID-19 treatment. Nano Today 36, 101019 (2021). https://doi.org/10.1016/j.nantod.2020.101019
X. Huang, N. Kong, X. Zhang, Y. Cao, R. Langer et al., The landscape of mRNA nanomedicine. Nat. Med. 28, 2273–2287 (2022). https://doi.org/10.1038/s41591-022-02061-1
Z. Tang, N. Kong, X. Zhang, Y. Liu, P. Hu et al., A materials-science perspective on tackling COVID-19. Nat. Rev. Mater. 5, 847–860 (2020). https://doi.org/10.1038/s41578-020-00247-y
Acknowledgements
This project was supported by the Natural Science Foundation of Fujian Province of China (No. 2022J01043) and China Scholarship Council (201806315005 and 201703170071).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest. They have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. Prof. Xingcai Zhang is an editorial board member for Nano-Micro Letters and was not involved in the editorial review or the decision to publish this article.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, X., Zhou, Q., He, Z. et al. ROS Balance Autoregulating Core–Shell CeO2@ZIF-8/Au Nanoplatform for Wound Repair. Nano-Micro Lett. 16, 156 (2024). https://doi.org/10.1007/s40820-024-01353-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40820-024-01353-0