
Abstract
In this work, we introduce a 2D materials family with chemical formula MX2 (M={As, Sb, Bi} and X={S, Se, Te}) having a rectangular 2D lattice. This materials family has been predicted by systematic ab-initio structure search calculations in two dimensions. Using density-functional theory and many-body perturbation theory, we study the structural, vibrational, electronic, optical, and excitonic properties of the predicted MX2 family. Our calculations reveal that the predicted SbX2 and BiX2 monolayers are stable while the AsX2 layers exhibit an in-plane ferroelectric instability. All materials display strong excitonic effects and good optical absorption within the infrared-to-visible range. Hence, these monolayers can harvest solar energy and serve in optoelectronics applications. Furthermore, our results indicate that exfoliation of the predicted MX2 monolayers from their bulk counterparts is experimentally viable.
Similar content being viewed by others

Introduction
In the last two decades, a remarkable array of two-dimensional (2D) materials has been successfully exfoliated from their bulk counterparts, including notable examples such as graphene1, transition-metal dichalcogenides (TMDCs)2, and MXenes3. These materials exhibit extraordinary properties4,5,6, positioning themselves as promising candidates for future optical, electronic, and mechanical devices. Consequently, the quest for 2D materials with a broad range of applications is at the forefront of materials design.
Recently, computational search methods have emerged as a viable approach for exploring archetypes of 2D materials7,8,9,10,11. Usually, the search starts with an existing material, with chemical combinatorics employed to design materials with the same prototype. This allows us to control the physical and chemical properties of the designed materials by tuning their composition. In addition, systematic exploration of different configurations (with a given stoichiometry) often leads to discovery of structures with lower energy than that of the starting configuration.
One of the most promising applications of the discovered 2D materials is the development of advanced optoelectronic devices4. Due to weak dielectric screening and confinement of electrons in two dimensions, the optical properties of 2D materials are often governed by strongly bound electron-hole pairs known as excitons12. These excitons can be harnessed and utilized for practical applications in next-generation optoelectronic and photonic devices such as light-emitting diodes12, solar cells and photodetectors13.
TMDCs14 and black phosphorus (BP)15 are among the most attractive 2D materials. They both display pronounced excitonic effects but have quite different optical properties otherwise. The absence of inversion symmetry in monolayer TMDCs leads to nonlinear optical properties and the presence of strong spin-orbit interactions. Locking of spin within the inequivalent K valleys leads to the ability to manipulate the valley degree of freedom using circularly polarized light14. Unlike TMDCs, BP is centrosymmetric and possesses a rectangular unit cell displaying in-plane anisotropy which results in an anisotropic optical absorption and emission spectrum15. Given the potential wide-ranging applications of 2D excitonic materials in next-generation optoelectronic devices, the search of 2D materials exhibiting strongly bound excitons and exceptional optical absorption across the infrared-to-visible range of the solar spectrum is very important.
In this paper, we predict a family of 2D materials based on first-principles density-functional theory (DFT) calculations, with stoichiometry MX2, where M = {As, Sb, Bi} and X = {S, Se, Te}, for promising applications in optoelectronic devices. Our state-of-the-art DFT and many-body perturbation theory calculations reveal that most of the predicted rectangular MX2 (r-MX2) monolayers are stable and absorb light within the infrared-to-visible range of the solar-energy spectrum and hence can harvest solar energy for optoelectronics applications. Furthermore, these monolayers possess inversion symmetry. They exhibit an indirect bandgap, a strong in-plane anisotropy, and strong excitonic effects. Since these monolayers have smaller exfoliation energy compared to the 1H-MoS2 monolayer (between 8–50% of the energy needed to exfoliate 1H-MoS2), and since their bulk counterparts have already been synthesized16,17,18, the two-dimensional exfoliation of the predicted r-MX2 monolayers seems experimentally viable.
Results and discussion
Motivation, structure, and stability
Motivation
We decided to study MX2 chalcogenides (M = {As, Sb, Bi} and X = {S, Se, Te}) because the whole set of materials should have similar properties (e.g., similar bonding patterns, electronic band structures, phonon dispersions, etc.), but offering the possibility to control them by changing the composition. For instance, the electronegativity decreases for the heavier elements in the MX2 series. Also, the heavier elements have more significant spin-orbit coupling (SOC), which usually plays a substantial role in tuning the bandgap.
The ratio M/X can provide considerable information about the exfoliation possibility in three-dimensional systems. Compounds of elements of the group V and VI of the periodic table, such as Bi2Se3 (M/X = 2/3), have an exfoliable bulk structure, consisting of sets of the so-called quintuple layers that are stacked together via vdW interactions. By increasing the ratio M/X = 1, a layered bulk phase is likely to be obtained, but it may no longer be exfoliable16,18. In contrast, for M/X = 0 (i.e., the bulk of pure S, Se, or Te, the system is no longer layered; it is composed of open or closed chains joined by vdW forces19. Therefore, we expected to find exfoliable 2D materials for a ratio of M/X = 1/2 and that we could use this MX2 stoichiometry to push our structural-search algorithm to a yet unexplored “playground” (we were unaware of the experimental identification of the reported BiS2 bulk phase16,18 when we started our structure search).
Ab-initio structure prediction
The predicted structure in this work was obtained by systematic ab-initio structure search calculations performed using the constrained minima hopping method (cMHM)20,21. This method employs an efficient dynamical algorithm, which couples DFT with short molecular dynamics simulations to explore the multidimensional potential energy surface (PES) of quasi-2D materials, intending to discover local and global minima on the PES22,23,24,25,26,27 (see Ref. 21 for technical details). In a previous work21, the cMHM successfully predicted several phases and reproduced the known 2D crystal phases of Bi monolayer. In this work, we perform similar structure search calculations as reported in Ref. 21 but for the BiS2 composition. In our structure search calculations, we considered one (3 atoms/cell) to eight (24 atoms/cell) formula units of BiS2. The obtained low-energy structures corresponding to local minima on PES were further optimized using tighter convergence criteria of k-mesh sampling and plane-wave energy cutoff (see Methods section) with the inclusion of SOC. The obtained lowest energy configuration was considered the ground state structure of BiS2 monolayer. This inference was further supported by a comparative energetics study of other candidate phases of BiS2 monolayers as discussed below.
Structural properties and crystallographic details
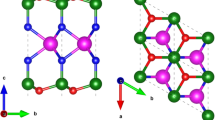
The obtained ground state configuration of the BiS2 monolayer, shown in Fig. 1, is centrosymmetric and belongs to the 2D layer group cm11 (no. 13); the 3D supercell containing vacuum along c axis belongs to the space group C2/m (no. 12). The conventional unit cell (shown in blue in Fig. 1) has a rectangular shape, whereas the primitive cell (shown in red in Fig. 1) has a rhombic shape. Following the conventional cell, the 2D Bravais lattice is rectangular and we refer to this system as the r-BiS2 monolayer. We have also calculated the relaxed geometries of the other rectangular pnictogen dichalcogenides to which we will refer as r-MX2 in the following.

a Top and (b) side views of the studied r-MX2 monolayer. The conventional (blue) and primitive (red) unit cells are drawn in panel (a).
The primitive lattice vectors of r-MX2 are a1 = (a/2, − b/2, 0) and a2 = (a/2, b/2, 0), as marked using red arrows in Fig. 1. The a lattice parameter varies from 14.5 Å for AsS2 to 16.7 Å for BiTe2. The values of b ranges between 3.7 Å for AsS2 and 5.5 Å for BiTe2. The angle between both vectors is ~ 28∘. Naturally, the heavier the constituent atoms are, the larger are the lattice parameters (see Fig. 2), but the effect is considerably more pronounced for the chalcogen atoms than for the pnictogen atoms.

a Calculated formation energy (Eform) of each r-MX2 monolayer (see Eq. (3)). b G0W0 (solid black line) and HSE06 band gap with (solid red line) and without (dashed line) spin-orbit coupling, and (c) lattice parameters a and b as defined in the text. In all panels the lines are a guide to the eye for systems with the same M (As, Sb, or Bi) atom.
Stability of r-MX2 monolayers
The calculation of elastic constants helps to verify the elastic and mechanical stability of the r-BiS2 monolayer28,29. Our calculated elastic constants (with SOC) are: C11 = 17.5, C12 = 6.9, C22 = 28.9, and C66 = 7.3 N/m. These elastic constants are of the same order of magnitude as in the buckled-hexagonal BiSb monolayer (C11 = C22 = 24.4, and C12 = 5.8 N/m)30. Dynamical stability of r-BiS2 and the other compounds has been checked by calculating the phonon dispersions which will be discussed in the section on vibrational properties below. Altogether, our calculations suggest that the predicted r-BiS2 monolayer is energetically, elastically, mechanically, and dynamically stable, providing support to the notion that it could be experimentally realized. The results on elastic constants and phonon dispersions of all r-MX2 monolayers studies in this manuscript are given in the Supplementary Table 1 and Supplementary Fig. 2.
In addition to the monolayer r-MX2 geometry predicted in this work, for the sake of comparison, we explicitly considered the two hexagonal TMDC-like structures, 1H-MX2 (trigonal prismatic) and 1T-MX2 (octahedral) geometries2. Our calculations reveal that the predicted r-MX2 structure is indeed the lowest-energy ground state geometry among all the studied geometries for the MX2 family; see Supplementary Fig. 2, which shows a systematic comparison of the relative energy among the three MX2 phases (1H, 1T, and rectangular). It is not surprising that the 1T- and 1H-MX2 geometries have relatively high energy: they both are highly-coordinated structures, and the MX2 compounds studied here seem to have an “incorrect” stoichiometry31 due to mixing of elements from the group V and VI in a proportion 1 to 2. Atoms S, Se, and Te already have 6 valence electrons, very close to reaching the octet19. Although this rule is not expected to be strictly followed, a significant deviation from it has to be energetically unfavorable which is manifested here in the relatively high energies of the hexagonal structures.
The SbS2, SbSe2, BiS2, and BiSe2 monolayers are already present in the Computational 2D Materials Database (C2DB)7 but only in the 1T-MX2 phase. The stability of these materials is labeled as “high”, except for the thermodynamic stability of BiSe2, which is “medium” (it lies relatively high in the minimum energy convex hull). As we mentioned before, the family of r-MX2 compounds has lower total energy compared to the 1T-MX2 geometry (i.e., the proposed phase happens to be energetically more stable).
The possibility of synthesizing the r-MX2 monolayer also requires a low formation energy Eform (see Eq. (3)). For instance, the 1H-MoS2 monolayer has been successfully exfoliated by micromechanical methods32 and has formation energy Eform ≈ 80 meV/atom33. Compared with all predicted r-MX2 monolayers, all formation energies are Eform ≲ 40 meV/atom, as shown in Fig. 2a. Since Eform is lower for all the orthorhombic MX2 compounds, its exfoliation from its bulk phase seems viable. In general, 2D materials with Eform < 200 meV/atom do not need a stabilizing substrate, and they are regarded stable as free-standing or suspended flakes34.
Experimentally, the bulk phase of r-BiS2 and r-BiSe2 (a layered monoclinic structure in space group C2/m) has been synthesized at ~ 5.5 GPa pressure16,17. The experimental atomic positions and lattice parameters are consistent with our calculations. There exist two other reports on bulk BiS2 obtained in similar growth conditions, but the crystal geometry was not specified18,35. Likely, it is the same monoclinic phase, which is the only phase of BiS2 reported so far. One of these studies indicates that bulk BiS2 is stable in water18. Overall, these studies suggest the possibility of synthesizing the two-dimensional r-BiS2 and r-BiSe2 through exfoliation, and a similar synthesis approach may be possible for the other r-MX2 monolayers.
Vibrational properties
Since the primitive unit cell of r-BiS2 monolayer contains six atoms (2 Bi and 4 S), there are 15 allowed optical phonon modes, which can be described by the following irreducible representation at the zone center
Here Ag and Bg modes are Raman active, whereas Au and Bu modes are infrared active. The predicted frequencies of these modes at the zone center are as follows: Ag = 34, 92, 173, 238, 282, and 469 cm−1; Au = 52 and 172 cm−1; Bg = 48, 103, and 215 cm−1; Bu = 92, 156, 210, and 291 cm−1.
It is worthwhile to have a closer look at the phonon dispersion of r-BiS2 shown in Fig. 3a. The dispersion has been calculated with a 2D Coulomb cutoff36 in order to prevent an LO/TO splitting which, otherwise, would occur at the Γ point for some of the phonon modes37. Instead of the LO/TO splitting, we observe an overbending at Γ of some of the infrared active modes. In particular the Au mode at 52 cm−1 displays an ultra-strong overbending in the Γ → C2 and the Γ → V2 directions (but not in the ΓY2 direction where the phonon displacement is perpendicular to the phonon wave vector). The origin of this overbending, marked by the red line in the dispersion, becomes clear when looking at the displacement pattern in panel (b): the mode is strongly polar with all sulfur atoms moving in one direction and all bismuth atoms moving in the opposite direction.

a Phonon dispersion of r-BiS2 monolayer calculated using density-functional perturbation theory with a 2D coulomb cutoff (see Supplementary Fig. 1 for calculated phonons of all r-MX2 monolayers studied in this work). Black circles denote the Raman active phonon modes and red lines depict the infrared active mode with overbending. b Vibration pattern for over-bending Au mode and for the two highest frequency Raman modes at Γ. c Off-resonant Raman spectrum at 300 K computed within DFPT.
The extreme overbending is actually connected to incipient ferroelectric behaviour. Actually, in the AsX2 compounds, this mode is soft (see Supplementary Fig. 2). Geometry relaxation along this mode leads to a polar structure of lower energy, displaying a dipole moment in the plane. The polar structures, along with the unit-cell parameters and the energy gain with respect to the non-polar structure are shown in the Supplementary Fig. 4. This adds the predicted materials to the recently published list of 2D ferroelectric materials, found by a computational high-throughput search38.
The highest phonon branch at 469 cm−1 is almost flat. This mode consist of bond-stretching of the (only) S-S bond in the structure as displayed in panel (b). All other atoms are at rest. The relatively large distance between neighboring S-S bonds explains the flatness of the band. Likewise, the second highest mode (291 cm−1 at Γ) is also very flat, because only sulfur atoms outside the S-S bond are participating, interacting only very loosely with the neighboring cluster of vibrating atoms.
The phonon dispersion of r-BiS2 does not have any soft modes and thus demonstrates the dynamical stability of this monolayer. The presence of minor imaginary phonon frequencies in the parabolic acoustic ZA branch near the zone center is ascribed to the numerical inaccuracies in the long-wavelength limit (q → 0)39,40 and can be observed in the calculated dispersion of many 2D materials. The calculated phonon dispersions of most of the r-MX2 family members (See Supplementary Fig. 2) are similar to that shown in Fig. 3, except for the r-AsX2 monolayers that exhibit a ferroelectric instability.
Figure 3c shows the computed non-resonant Raman spectrum of r-BiS2. It demonstrates that 7 out of the total of 9 Raman active modes are clearly visible in the spectrum. We thus expect that Raman spectroscopy will be a very useful tool to identify r-MX2 monolayers and to distinguish them from hexagonal phases of 2D materials (which only have two Raman active modes at high energy and the layer shear mode at very low energy41).
Electronic properties
Before showing electronic band structure results from our DFT calculations, let us briefly discuss what could be expected from the predicted r-MX2 monolayers, in contrast to the 1H- and 1T-MX2 monolayers. The 1H- and 1T-MX2 monolayer structures proposed so far are metallic7 – and hence are not suitable for optoelectronics applications – for a straightforward reason: they have an odd number of electrons per unit cell. In our case, the rhombic lattice of r-MX2 (Fig. 1) doubles the unit cell, removing that band-filling constraint and, thus, allowing a bandgap at the Fermi energy.
Figure 4 shows the electronic band structures of all r-MX2 monolayers calculated using G0W042,43,44 (solid lines) and PBE45 (dashed lines) methods with the inclusion of SOC effects. We notice that the overall dispersion of bands does not change substantially as a function of the employed exchange-correlation (XC) functional. However, the bandgap varies considerably for different employed XC functionals (see Table 1). While the effect of SOC is almost negligible for the lighter elements considered, it can be substantial for the heavier constituent elements, especially for the BiX2 series. Note, there is no SOC-induced spin splitting of bands owing to the centrosymmetric structure of the r-MX2 monolayers.

Electronic band structure of r-MX2 monolayers calculated using G0W0 (solid lines) and PBE (dashed lines) including the effects of spin-orbit coupling. The k-path for BiS2 is formed by Γ (0.0, 0.0), C (−0.268, 0.268), C2 (0.268, 0.732), Y2 (0.5, 0.5), and V2 (0.0, 0.5). The coordinates for C, C2, and Y2 depend on the cell parameters and were determined by the materialscloud’s SeeK-path tool64. The Fermi energy (EF) is set at the valence band maximum.
Figure 2b and Table 1 show the bandgap calculated using different theoretical methods. Most of the r-MX2 monolayers are predicted to absorb light within the visible range of the solar spectrum. An indirect bandgap is observed in As and Sb monolayers while a direct gap is seen in Bi monolayers. This could be a significant advantage compared to the traditional TMDCs as it strongly suppresses the electron-hole recombination (luminescence) during the optoelectronic applications. Recent works on TMDCs report that non-radiative electron-hole recombination channels can be further suppressed by electrostatic doping46 and strain47. We anticipate that similar approaches can be applied to enhance the optoelectronic performance of the predicted r-MX2 monolayers.
Optical properties and many-body effects
In order to obtain the absorption spectrum, including electron-hole interactions, we solve the Bethe-Salpeter equation (BSE)48 on top of the G0W0 calculations within the Tamm-Dancoff approximation42 using the YAMBO code43. The optical absorption spectrum along the polarization direction μ is described by the imaginary part of the 2D polarizability tensor and is given by48
where A is the area of the unit cell, vμ is component of the velocity operator along the direction μ, \({A}_{vc{{{\bf{k}}}}}^{S}\) are the expansion coefficients of the exciton eigenstate S in the electron-hole basis and ES is the exciton energy.
The optical absorption spectra for r-MX2 monolayers obtained by solving the BSE are shown in Fig. 5. The solid and dashed colored lines show the absorption spectrum computed with and without including electron-hole interactions. The onset of direct optical absorption occurs within the infrared-to-visible range of the solar spectrum. Moreover, for all the monolayers, the absorption onset starts before the indirect gap. The double peak structure (A/B excitons) observed in 2H-Mo(W)X2 monolayers2 due to spin-orbit splitting of the top valence band is absent in these materials due to the inversion symmetry of the crystal.

Optical absorption spectra for different r-MX2 monolayers. The solid and dashed lines correspond to the optical absorption spectrum calculated with and without considering electron-hole interactions. The red and blue lines in each subplot represent the absorption spectrum calculated along x and y polarization directions, respectively. The x and y directions are shown in Fig. 1. All spectra are plotted with a linear broadening ranging from 10 meV to 100 meV with the minimum and maximum energy. The solid and dashed vertical lines in each subplot denote the computed energies (with SOC effects) of the direct gap and the bandgap, respectively.
In Fig. 5, we also observe that strongly bound excitons dominate the optical absorption spectrum for all r-MX2 monolayers. We find that for all the studied monolayers, the absorption onset shifts by a few hundred meV when excitonic effects are included, which indicates that excitons possess large exciton binding energies. Similar to the case of excitons in MoS2 and other transition metal dichalcogenides14,41, these large binding energies are due to the reduced environmental screening in monolayers and the quasi-2D nature of the excitons.
The dielectric screening increases when going down the periodic table for the chalcogen atoms, which is in line with a decrease in direct bandgap, as shown in Fig. 5. The lowering of direct bandgap results in a red shift of absorption onset with an increase in the atomic number of the chalcogen atom, as shown in Table 2. In contrast, we do not observe a clear trend with the pnictogen atomic number. The bandgaps and the first exciton energies are highest for the Sb-based compounds.
Finally, we look at the anisotropic optical absorption of these materials. We start by comparing the excitons in MX2 monolayers with monolayer BP as both materials exhibit anisotropy and possess inversion symmetry. Due to the difference in crystal symmetries for MX2 monolayer (C2h) and monolayer BP (D2h), we observe different optical selection rules. In monolayer BP, the bright excitons transform under two different representations (B2u and B3u), which results in selective coupling of excitons to either x or y polarized light49. In contrast, the bright excitons in r-MX2 monolayers transform according to the single representation (Bu) which can couple to both x or y polarized light (yet, with different intensities as the calculations show). In Fig. 5, we present the absorption spectra for all the monolayers along the x and y directions. (The x and y directions are shown in Fig. 1). The first few bound excitons in all monolayers exhibit a greater oscillator strength along the y direction than the x direction. This anisotropy in the optical absorption spectrum arises from the in-plane geometric anisotropy within the crystal structure.
Role of temperature
In general, an increase in temperature leads to a decrease in the band gap of semiconductors, primarily due to electron-phonon interactions. This effect is observed not only in traditional semiconductors but also in two-dimensional systems like MX2 compounds, which include materials such as MoS2, WSe2, and WS250,51. These changes in the band gap result in a slight redshift (kBT ~ 26 meV) in the optical absorption spectra, including the energy peaks associated with excitons (as depicted in Fig. 5). Similar to the band gap, the size of the exciton peaks diminishes with increasing temperature, although they remain experimentally detectable in materials like MoSe2.
MoSe2, with its comparable electronic band gap (~2 eV) and optical properties50, provides a valuable point of reference for comparing and extrapolating the temperature-induced effects observed in our compounds. Consequently, at room temperature, we anticipate only minor or negligible perturbations in the band gap and, consequently, in the optical and exciton properties.
Outlook
Compared to other known phases of MX2 monolayers, such as the 1H- and 1T-MX2 geometries, our predicted r-MX2 monolayers exhibit the highest stability, as indicated by their lowest formation energy. These materials can be easily exfoliated from their bulk counterparts, requiring only a tiny amount of energy (8–50% of the energy needed to exfoliate 1H-MoS2 monolayer). Some of the r-MX2 monolayers in our study are indirect bandgap semiconductors. The bandgaps predicted by the G0W0 method range from 0.83 eV in BiTe2 to 2.24 eV in SbS2. Importantly, all the r-MX2 monolayers absorb light within the infrared-to-visible range of the solar spectrum and exhibit significant excitonic effects with large exciton binding energies. Our findings suggest promising applications of r-MX2 monolayers in optoelectronic devices operating in the solar-energy range. Furthermore, considering that the three-dimensional counterpart of r-BiS2 has already been experimentally synthesized, and the formation energy of r-MX2 monolayers is lower than half that of 1H-MoS2 monolayers, the exfoliation of r-MX2 monolayers appears to be a feasible experimental approach.
Methods
DFT details
All DFT calculations were performed using the Vienna ab initio simulation package (VASP)52 and the Quantum ESPRESSO (QE) package53 with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation (XC) functional45. We used fully relativistic projector augmented wave54,55 for VASP calculations and norm-conserving pseudopotentials from the PseudoDojo project56 for the QE calculations. Spin-orbit coupling (SOC) was included in all DFT calculations unless specified otherwise.
In order to obtain the equilibrium lattice parameters, we first performed structural relaxation with the VASP code. We later performed self-consistent calculations to obtain the ground state charge densities. A plane wave energy cutoff of 400 eV and a uniform Γ centred k-point sampling of 24 × 24 × 1 was employed for all the VASP calculations. We calculated the electronic band structures using screened-hybrid functional of Heyd-Scurseria-Ernzerhof (HSE06)57 as PBE-XC functional45 generally underestimates the bandgap. PyProcar58 and MechElastic29 packages were used for further analysis of the electronic and elastic properties, respectively.
Formation energies for all the materials were computed using the following expression
where E2D/3D are monolayer and bulk energies of MX2 and N2D/3D are the number of atoms in the unit cell33,34,59. This energy compares the energetics of the monolayer and bulk structures, which allows us to judge the possible experimental realization by means of exfoliation.
We employed density-functional perturbation theory (DFPT)60 for phonon calculations as implemented within the QE code. All the structures were relaxed again using the QE code with a convergence threshold of 10−6 Ry and 10−5 Ry/Bohr for total energy and forces, respectively. To obtain the ground state charge densities, we used a plane wave cutoff of 100 Ry and a uniform Γ centred 9 × 9 × 1 k-point grid. We set a vacuum spacing of 16Å for all materials and employed a 2D Coulomb cutoff technique36 to remove spurious interactions along the out-of-plane direction. In order to obtain phonon dispersions, we computed dynamical matrices on a coarse 4 × 4 × 1 Γ centred q-point grid. These dynamical matrices were Fourier interpolated to obtain the force-constant matrix in real space, which in turn is employed to calculate the phonon dispersion on a fine q-point path.
For the GW calculations, we first obtained the Kohn-Sham energies and wave functions on a uniform 9 × 9 × 1 k-point grid by performing a non self-consistent calculation with a plane wave cutoff of 80 Ry using the QE code. In order to construct the microscopic dielectric tensor, we used a plane wave cutoff of 8 Ry and performed the summation with 1000 Kohn-Sham states. The frequency dependence of the dielectric tensor was described within a plasmon-pole approximation61. A 2D Coulomb cutoff along the out-of-plane was employed to remove the interactions with periodic images. In order to speed up the convergence of G0W0 calculations with respect to bands and k-points, we used a G-terminator62 and RIM-W63 technique, respectively.
To obtain well-converged absorption spectra, we used a uniform Γ-centred 30 × 30 × 1 k-point grid for all r-MX2 monolayers. A total of 250 bands and a cut-off of 4 Ry were used to build the static dielectric tensor. We included the top eight valence and the bottom eight conduction bands to construct the BSE interaction kernel. A plane-wave cutoff of 60 Ry and 4 Ry was used in the construction of bare exchange and screened Coulomb blocks, respectively.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Additional data on elastic constants and stability in comparison to hexagonal phases, as well as all calculated phonon dispersions are available in the Supplementary Table 1 and Supplementary Fig. 2 accompanying this paper. Input and output files of our DFT, GW, and BSE calculations on band-structures, optical spectra and phonon dispersions are available in the NOMAD repository with identifier https://doi.org/10.17172/NOMAD/2024.01.03-1. All relevant data is also available from the authors upon reasonable request.
Code availability
The first-principles DFT calculations were performed using the privately-licensed VASP, and the open-source Quantum ESPRESSO software.
References
Novoselov, K. S. et al. Two-dimensional gas of massless dirac fermions in graphene. Nature 438, 197–200 (2005).
Mak, K. F., Lee, C., Hone, J., Shan, J. & Heinz, T. F. Atomically thin mos2: a new direct-gap semiconductor. Phys. Rev. Lett. 105, 136805 (2010).
Naguib, M., Mochalin, V. N., Barsoum, M. W. & Gogotsi, Y. 25th anniversary article: MXenes: a new family of two-dimensional materials. Adv. Mater. 26, 992–1005 (2014).
Tan, T., Jiang, X., Wang, C., Yao, B. & Zhang, H. 2d material optoelectronics for information functional device applications: status and challenges. Adv. Sci. 7, 2000058 (2020).
Sangwan, V. K. & Hersam, M. C. Electronic transport in two-dimensional materials. Annu. Rev. Phys. Chem. 69, 299–325 (2018).
Akinwande, D. et al. A review on mechanics and mechanical properties of 2d materials-graphene and beyond. Extreme Mech. Lett. 13, 42–77 (2017).
Haastrup, S. et al. The computational 2d materials database: high-throughput modeling and discovery of atomically thin crystals. 2D Mater. 5, 042002 (2018).
Lyngby, P. & Thygesen, K. S. Data-driven discovery of 2D materials by deep generative models. npj Comput. Mater. 8, 232 (2022).
Zhou, J. et al. 2dmatpedia, an open computational database of two-dimensional materials from top-down and bottom-up approaches. Sci. Data 6, 1–10 (2019).
Mounet, N. et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nat. Nanotechnol. 13, 246–252 (2018).
Wang, H.-C., Schmidt, J., Marques, M. A. L., Wirtz, L. & Romero, A. H. Symmetry-based computational search for novel binary and ternary 2D materials. 2D Mater. 10, 035007 (2023).
Xiao, J., Zhao, M., Wang, Y. & Zhang, X. Excitons in atomically thin 2d semiconductors and their applications. Nanophotonics 6, 1309–1328 (2017).
Mueller, T. & Malic, E. Exciton physics and device application of two-dimensional transition metal dichalcogenide semiconductors. Npj 2D Mater. Appl. 2, 29 (2018).
Wang, G. et al. Colloquium: excitons in atomically thin transition metal dichalcogenides. Rev. Mod. Phys. 90, 021001 (2018).
Wang, X. et al. Highly anisotropic and robust excitons in monolayer black phosphorus. Nat. Nanotechnol. 10, 517–521 (2015).
Yamamoto, A., Hashizume, D., Bahramy, M. S. & Tokura, Y. Coexistence of monochalocogen and dichalocogen ions in bise2 and bis2 crystals prepared at high pressure. Inorg. Chem. 54, 4114–4119 (2015).
Kevy, S. M. et al. Investigation of the high pressure phase bis2: Temperature-resolved structure and compression behavior to 60 gpa. J. Alloys Compd. 789, 588–594 (2019).
Silverman, M. S. High pressure synthesis of new compounds–bismuth diselenide and bismuth monosulfide monoselenide. Inorg. Chem. 4, 587–588 (1965).
Hess, P. Bonding, structure, and mechanical stability of 2d materials: the predictive power of the periodic table. Nanoscale Horiz. 6, 856–892 (2021).
Eivari, H. A. et al. Two-dimensional hexagonal sheet of tio2. Chem. Mater. 29, 8594–8603 (2017).
Singh, S. et al. Low-energy phases of bi monolayer predicted by structure search in two dimensions. J. Phys. Chem. Lett. 10, 7324–7332 (2019).
Goedecker, S. Minima hopping: an efficient search method for the global minimum of the potential energy surface of complex molecular systems. J. Chem. Phys. 120, 9911–9917 (2004).
Amsler, M. & Goedecker, S. Crystal structure prediction using the minima hopping method. J. Chem. Phys. 133, 224104 (2010).
Amsler, M. K. Crystal structure prediction based on density functional theory. Ph.D. thesis, University of Basel (2014).
Singh, S., Ibarra-Hernández, W., Valencia-Jaime, I., Avendaño-Franco, G. & Romero, A. H. Investigation of novel crystal structures of bi–sb binaries predicted using the minima hopping method. Phys. Chem. Chem. Phys. 18, 29771–29785 (2016).
Pavlic, O. et al. Design of mg alloys: the effects of li concentration on the structure and elastic properties in the mg-li binary system by first principles calculations. J. Alloys Compd. 691, 15–25 (2017).
Singh, S. K. Structural Prediction and Theoretical Characterization of Bi-Sb Binaries: Spin-Orbit Coupling Effects. Ph.D. thesis, West Virginia University (2018).
Maździarz, M. Comment on ‘the computational 2d materials database: high-throughput modeling and discovery of atomically thin crystals’. 2D Mater. 6, 048001 (2019).
Singh, S. et al. Mechelastic: a python library for analysis of mechanical and elastic properties of bulk and 2d materials. Comput. Phys. Commun. 267, 108068 (2021).
Singh, S. & Romero, A. H. Giant tunable rashba spin splitting in a two-dimensional bisb monolayer and in bisb/aln heterostructures. Phys. Rev. B 95, 165444 (2017).
Brazhkin, V. V., Dyuzheva, T. I. & Zibrov, I. P. New pressure-induced phase transitions in bismuthinite. JETP Lett. 114, 470–474 (2021).
Novoselov, K. et al. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. U.S.A. 102, 10451–10453 (2005).
Zhuang, H. L. & Hennig, R. G. Single-layer group-iii monochalcogenide photocatalysts for water splitting. Chem. Mater. 25, 3232–3238 (2013).
Singh, A. K., Mathew, K., Zhuang, H. L. & Hennig, R. G. Computational screening of 2d materials for photocatalysis. J. Phys. Chem. Lett. 6, 1087–1098 (2015).
Adapa, V., Ramakrishnan, A., Heinz, N. & Dinda, G. Microstructural evolution during laser metal deposition of bismuth chalcogenides (bi2se3 and bi2te3). J. Alloys Compd. 774, 509–514 (2019).
Sohier, T., Calandra, M. & Mauri, F. Density functional perturbation theory for gated two-dimensional heterostructures: theoretical developments and application to flexural phonons in graphene. Phys. Rev. B 96, 075448 (2017).
Sohier, T., Gibertini, M., Calandra, M., Mauri, F. & Marzari, N. Breakdown of optical phonons’ splitting in two-dimensional materials. Nano Lett. 17, 3758–3763 (2017).
Kruse, M. et al. Two-dimensional ferroelectrics from high throughput computational screening. Npj Comput. Mater. 9, 1–11 (2023).
Royo, M., Hahn, K. R. & Stengel, M. Using high multipolar orders to reconstruct the sound velocity in piezoelectrics from lattice dynamics. Phys. Rev. Lett. 125, 217602 (2020).
Royo, M. & Stengel, M. Exact long-range dielectric screening and interatomic force constants in quasi-two-dimensional crystals. Phys. Rev. X 11, 041027 (2021).
Molina-Sánchez, A., Hummer, K. & Wirtz, L. Vibrational and optical properties of MoS2: from monolayer to bulk. Surf. Sci. Rep. 70, 554–586 (2015).
Benedict, L. X., Shirley, E. L. & Bohn, R. B. Theory of optical absorption in diamond, si, ge, and gaas. Phys. Rev. B 57, R9385–R9387 (1998).
Sangalli, D. et al. Many-body perturbation theory calculations using the yambo code. J. Phys. Condens. Matter 31, 325902 (2019).
Marini, A., Hogan, C., Grüning, M. & Varsano, D. yambo: an ab initio tool for excited state calculations. Comput. Phys. Commun. 180, 1392–1403 (2009).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Lien, D.-H. et al. Electrical suppression of all nonradiative recombination pathways in monolayer semiconductors. Science 364, 468–471 (2019).
Kim, H., Uddin, S. Z., Higashitarumizu, N., Rabani, E. & Javey, A. Inhibited nonradiative decay at all exciton densities in monolayer semiconductors. Science 373, 448–452 (2021).
Rohlfing, M. & Louie, S. G. Electron-hole excitations and optical spectra from first principles. Phys. Rev. B 62, 4927–4944 (2000).
Tran, V., Fei, R. & Yang, L. Quasiparticle energies, excitons, and optical spectra of few-layer black phosphorus. 2D Mater. 2, 044014 (2015).
Liu, H.-L. et al. Temperature-dependent optical constants of monolayer mos2, mose2, ws2, and wse2: spectroscopic ellipsometry and first-principles calculations. Sci. Rep. 10, 15282 (2020).
Choi, B. K. et al. Temperature dependence of band gap in mose2 grown by molecular beam epitaxy. Nanoscale Res. Lett. 12, 1–7 (2017).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Giannozzi, P. et al. Advanced capabilities for materials modelling with quantum ESPRESSO. J. Phys. Condens. Matter 29, 465901 (2017).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
van Setten, M. et al. The pseudodojo: training and grading a 85 element optimized norm-conserving pseudopotential table. Comput. Phys. Commun. 226, 39–54 (2018).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Erratum: “hybrid functionals based on a screened coulomb potential" [j. chem. phys. 118, 8207 (2003)]. J. Chem. Phys. 124, 219906 (2006).
Herath, U. et al. Pyprocar: a python library for electronic structure pre/post-processing. Comput. Phys. Commun. 251, 107080 (2020).
Zhuang, H. L. & Hennig, R. G. Computational search for single-layer transition-metal dichalcogenide photocatalysts. J. Phys. Chem. C 117, 20440–20445 (2013).
Baroni, S., de Gironcoli, S., Dal Corso, A. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515–562 (2001).
Godby, R. W. & Needs, R. J. Metal-insulator transition in kohn-sham theory and quasiparticle theory. Phys. Rev. Lett. 62, 1169–1172 (1989).
Bruneval, F. & Gonze, X. Accurate GW self-energies in a plane-wave basis using only a few empty states: towards large systems. Phys. Rev. B 78, 085125 (2008).
Guandalini, A., D’Amico, P., Ferretti, A. & Varsano, D. Efficient gw calculations in two dimensional materials through a stochastic integration of the screened potential. npj Comput. Mater. 9, 44 (2023).
Hinuma, Y., Pizzi, G., Kumagai, Y., Oba, F. & Tanaka, I. Band structure diagram paths based on crystallography. Comput. Mater. Sci. 128, 140–184 (2017).
Acknowledgements
This research was funded in part, by the Luxembourg National Research Fund (FNR), Inter Mobility 2DOPMA, grant reference 15627293. For the purpose of open access, the authors have applied a Creative Commons Attribution 4.0 International (CC BY 4.0) license to any Author Accepted Manuscript version arising from this submission. This work was also partially supported by Fondecyt Grants No. 1191353, 1231487, 1220715. J.D.M. was funded by the National Agency of Research and Development (ANID) through grants Fondecyt postdoctorado number 3200697 and Fondecyt regular number 1230747. F.M. was funded by the Center for the Development of Nanoscience and Nanotechnology CEDENNA AFB220001, and from Conicyt PIA/Anillo ACT192023. This research was partially supported by the supercomputing infrastructure of the NLHPC (ECM-02). We also acknowledge the use of University of Luxembourg high-performance computing (ULHPC) and computational resources awarded by XSEDE, a project supported by National Science Foundation grant number ACI-1053575. The authors also acknowledge the support from the Texas Advances Computer Center (with the Stampede2 and Bridges supercomputers). We also acknowledge the Super Computing System (Thorny Flat) at WVU, which is funded in part by the National Science Foundation (NSF) Major Research Instrumentation Program (MRI) Award #1726534, and West Virginia University. AHR also recognizes the support of West Virginia Research under the call research challenge grand program 2022 and NASA EPSCoR Award 80NSSC22M0173. KMR acknowledges support from the Office of Naval Research Grant N00014-21-1-2107. SS was supported by the U.S. Department of Energy, Office of Science, Office of Fusion Energy Sciences, Quantum Information Science program under Award Number DE-SC-0020340. SS also acknowledges support from the University Research Awards at the University of Rochester.
Author information
Authors and Affiliations
Contributions
This project was conceived by S.S. and the reported 2D materials family were predicted by structure search calculations conducted by S.S. J.D.M., F.M., K.M.R., and A.H.R. studied the electronic structure and stability of the reported monolayers. M.N. and L.W. performed calculations to study the optical and vibrational properties. All authors discussed the findings. The manuscript was written by J.D.M., M.N., F.M., L.W., and S.S. All authors read and commented on the manuscript. J.D.M. and M.N. equally contributed to this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mella, J.D., Nalabothula, M., Muñoz, F. et al. Prediction of BiS2-type pnictogen dichalcogenide monolayers for optoelectronics. npj 2D Mater Appl 8, 4 (2024). https://doi.org/10.1038/s41699-023-00439-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41699-023-00439-4
