
Abstract
Split-hand/foot malformation (SHFM) shows diverse heterogeneity and manifests with reduced penetrance and variable expressivity. This study investigated the underlying genetic cause of a family segregating SHFM. Exome sequencing followed by Sanger sequencing identified a novel single nucleotide heterozygous variant (NC_000019.9 (NM_005499.3):c.1118del) in UBA2 cosegregating in the family in an autosomal dominant manner. Our findings conclude that reduced penetrance and variable expressivity are the two remarkable and unusual features of SHFM.
Similar content being viewed by others

Data Report
Split-hand/foot malformation (SHFM; OMIM: 183600) or ectrodactyly is a rare congenital disorder that comprises a broad spectrum of limb abnormalities with variable phenotypes1. SHFM demonstrates a significant amount of clinical heterogeneity, ranging from a mild phenotype in the form of the shortening of a single central digit to a severe phenotype of monodactyly, and displays various modes of inheritance with variable expressivity and reduced penetrance2,3.
Herein, we report the clinical genetic analysis of a Pakistani family with two affected sisters with ectrodactyly/SHFM and variable types of other skeletal anomalies, including clinodactyly and brachydactyly. Peripheral whole blood samples were obtained after informed consent from two affected (III-1 and III-3) and one unaffected siblings (III-2) and both parents (II-1 and II-2; Fig. 1i). The Institutional Review Board (IRB) and Ethics Committees of The University of Central Punjab, Lahore, Pakistan approved the current study. The mother (II-2) of the female patients (III-1, III-3) was phenotypically asymptomatic (Fig. 1ii a–e). The affected girl (III-1) was 25 years old at the time of the study. She presented bilateral ectrodactyly of both hands and unilateral ectrodactyly in the right foot. Photographs and radiographs revealed an aplasia (absence) of the third finger of the left hand and complete aplasia of the third and fourth fingers with metacarpals of the right hand. Additionally, a radiograph of the left hand showed a small hypoplastic bone at the fourth metacarpal, and the second and third metacarpals were fused. In addition, clinodactyly of the second finger of the left hand could be seen in the radiographs. Furthermore, unilateral ectrodactyly in the right foot in the form of complete aplasia of the middle toe with an absence of the central metatarsal as well as a partial fusion of the fourth and fifth metatarsal bones were observed for the right foot (Fig. 1ii, f–j). The second affected girl (III-3) was 27 years old at the time of this study and presented with unilateral ectrodactyly of the right hand. However, she had no ectrodactyly in her feet. Photographs and radiographs revealed polydactyly with syndactyly in the right hand and clinodactyly of the fourth and fifth fingers of the right hand and the third and fifth fingers of the left hand. In addition, the middle finger of the left hand was short, with an absent distal phalanx. Moreover, aplasia of the terminal phalanx of the third toe of the right foot and deviated toes of both feet were notable; however, it was without dislocation (Fig. 1ii, k–o).

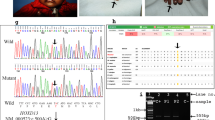
(i) Pedigree diagram showing the segregation of the UBA2 variant in an autosomal dominant manner. Male and female members are shown by squares and circles, respectively. Filled symbols denote the affected participants. The crossed shapes show the deceased individuals. The symbols labeled with asterisks designate the participants of the study. The genotypes are shown below each participant’s symbol. (ii) a–e The asymptomatic presentation of the mother of the affected individuals. f Facial picture of patient III-1 showing no facial dysmorphism. g, h Bilateral ectrodactyly of both hands and unilateral ectrodactyly in the right foot of patient III-1. i, j Radiographs of the hands and feet, respectively, of the patient (III-1). k Facial photograph of patient III-3 with no dysmorphic features. l, m Unilateral ectrodactyly of the right hand; however, there was no ectrodactyly in her feet. n, o Radiographs of the hands and feet, respectively. (iii) A The typical UBA2 gene structure consists of 17 exons. The variant is located in exon 11. B Sanger sequencing chromatograms of the unaffected father (II-1), asymptomatic heterozygous mother (II-2), an affected individual (III-1), an unaffected brother (III-2), and another affected member (III-3) in the pedigree.
Exome sequencing of the DNA from the affected sisters was performed to identify the causal genetic variant. Exome sequencing, filtering strategy and validation/cosegregation of the candidate variants are described previously elsewhere3. Upon analysis, we identified a heterozygous 1-bp deletion (NC_000019.9 (NM_005499.3): c.1118del) in exon 11 of the UBA2 gene residing on chromosome 19q13.11 (Fig. 1iii A). The variant is predicted to affect the frameshift of the UBA2 gene by creating a premature stop codon (NM_005499.3 (NP_005490.1):p.(Arg373Asnfs*21) and may lead to nonsense-mediated decay (NMD) of the truncated transcripts. Both affected sisters (III-1 and III-3) were heterozygous (WT/Mut) for this 1-bp deletion (Fig. 1iii B III-1, III-3). It was absent in the unaffected father (II-1) and unaffected sibling (III-2) (Fig. 1iii B II-1, III-2); however, the phenotypically unaffected mother (II-2) was heterozygous for this deletion (Fig. 1iii B II-2). The absence of ectrodactyly/SHFM and other clinical symptoms associated with UBA2 mutations in the unaffected mother may be due to incomplete penetrance, especially in autosomal dominant genetic disorders. Patients harboring a disease-causing variant remain asymptomatic throughout their life. According to ACMG and AMP guidelines4, the UBA2 variant is predicted to be pathogenic (PVS1, PM2, PP1, PP4), with a CADD score of 25.5. Additionally, this variant is absent in gnomAD. UBA2 had a high pLI (1.0) score (probability of loss-of-function intolerance). The pLI score ranges from 0 (most tolerant) to 1 (most intolerant) for genes containing loss-of-function mutations5.
The affected individuals in this report showed significant phenotypic variability and reduced penetrance. This is in line with typical SHFM features, as demonstrated by individuals with chromosome 19q13.11 microdeletion syndrome (encompassing the UBA2 gene) and point mutations in the UBA2 gene6,7,8. UBA2 (ubiquitin-like modifier-activating enzyme 2, OMIM *613295) is highly expressed in the developing limb buds of mice9. The UBA2 ortholog (uba2) in zebrafish was found to be expressed in the eye, brain, and pectoral fins. Furthermore, uba2-null fish were found to have low growth, microphthalmia, microcephaly, abnormal fins, and mandibular hypoplasia6. UBA2 is composed of 17 coding exons and encodes a protein of 640 amino acids. According to HGMD® Professional 2022.2, there are only five reports of 15 pathogenic variants in UBA2. Recently, a heterozygous frameshift (c.327delT, p. Phe109Leufs*3) variant in UBA2 was reported by Wang et al. in a boy and his mother. The affected boy had aplasia cutis congenita, bilateral SHFM/ectrodactyly, and other anomalies. However, the mother, also heterozygous for the same variant, had aplasia cutis congenita but was otherwise healthy, suggesting incomplete or reduced penetrance10. In conclusion, the current study reports a novel frameshift variant in UBA2 in a Pakistani family with SHFM. Our findings extend the phenotypic spectrum and allelic heterogeneity of UBA2-related phenotypes and further emphasize the vital role of UBA2 in limb development.
Data
The variant data are submitted to ClinVar, and an accession number (SCV002754416) has been assigned.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3292.
References
Duijf, P. H., van Bokhoven, H. & Brunner, H. G. Pathogenesis of split-hand/split-foot malformation. Hum. Mol. Genet. 12, R51–R60 (2003).
Gurrieri, F. & Everman, D. B. Clinical, genetic, and molecular aspects of split-hand/foot malformation: an update. Am. J. Med. Genet. A 161a, 2860–2872 (2013).
Spielmann, M. et al. Exome sequencing and CRISPR/Cas genome editing identify mutations of ZAK as a cause of limb defects in humans and mice. Genome Res. 26, 183–191 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Schnur, R. E. et al. UBA2 variants underlie a recognizable syndrome with variable aplasia cutis congenita and ectrodactyly. Genet. Med. 23, 1624–35. (2021).
Abe, K. T. et al. 19q13.11 microdeletion: clinical features overlapping ectrodactyly ectodermal dysplasia-clefting syndrome phenotype. Clin. Case Rep. 6, 1300–07. (2018).
Aerden, M. et al. Genotype-phenotype correlations of UBA2 mutations in patients with ectrodactyly. Eur. J. Med. Genet. 63, 104009 (2020).
Costa, M. W. et al. Complex SUMO-1 regulation of cardiac transcription factor Nkx2-5. PLoS ONE 6, e24812 (2011).
Wang, Y., Dupuis, L., Jobling, R. & Kannu, P. Aplasia cutis congenita associated with a heterozygous loss-of-function UBA2 variant. Br. J. Dermatol. 182, 792–94. (2020).
Acknowledgements
We thank the family members who participated in this study and the medical staff at local general hospitals. We are thankful to Prof. Reiner Siebert, Institute of Human Genetics, Ulm University and Ulm University Medical Center, 89081, Ulm, Germany, for his support during the study.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.A. and N.W. Data curation: A.P., N.W. Methodology and Formal analysis: N.W., S.A.K., A.P. Clinical investigations: A.P., M.T., N.K. Supervision: N.W., A.A. Writing—original draft: A.P., S.A.K., N.K. Writing—review & editing: M.T., S.A.K., N.K., and N.W.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Parveen, A., Tariq, M., Khan, S.A. et al. A novel frameshift variant in UBA2 causing split-hand/foot malformations in a Pakistani family. Hum Genome Var 10, 16 (2023). https://doi.org/10.1038/s41439-023-00242-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-023-00242-z
