当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Shrinking Self-Interaction Errors with the Fermi–Löwdin Orbital Self-Interaction-Corrected Density Functional Approximation
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2018-11-09 00:00:00 , DOI: 10.1021/acs.jpca.8b09940 Kamal Sharkas , Lin Li 1 , Kai Trepte , Kushantha P. K. Withanage , Rajendra P. Joshi , Rajendra R. Zope 2 , Tunna Baruah 2 , J. Karl Johnson 1 , Koblar A. Jackson , Juan E. Peralta
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2018-11-09 00:00:00 , DOI: 10.1021/acs.jpca.8b09940 Kamal Sharkas , Lin Li 1 , Kai Trepte , Kushantha P. K. Withanage , Rajendra P. Joshi , Rajendra R. Zope 2 , Tunna Baruah 2 , J. Karl Johnson 1 , Koblar A. Jackson , Juan E. Peralta
Affiliation
|
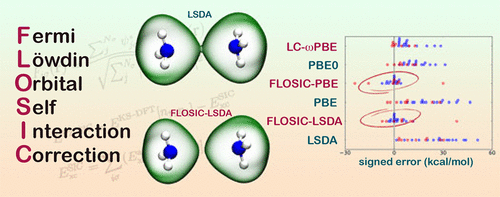
The self-interaction error (SIE) is one of the major drawbacks of practical exchange-correlation functionals for Kohn–Sham density functional theory. Despite this, the use of methods that explicitly remove SIE from approximate density functionals is scarce in the literature due to their relatively high computational cost and lack of consistent improvement over standard modern functionals. In this article we assess the performance of a novel approach recently proposed by Pederson, Ruzsinszky, and Perdew [J. Chem. Phys.2014, 140, 121103] for performing self-interaction free calculations in density functional theory based on Fermi orbitals. To this end, we employ test sets consisting of reaction energies that are considered particularly sensitive to SIE. We found that the parameter-free Fermi–Löwdin orbital self-interaction correction method combined with the standard local spin density approximation (LSDA) and Perdew–Burke–Ernzerhof (PBE) functionals gives a much better estimate of reaction energies compared to their parent LSDA and PBE functionals for most of the reactions in these two sets. They also perform on par with the global PBE0 and range-separated LC-ωPBE hybrids, which partially eliminate the SIE by including Hartree–Fock exchange. This shows the potential of the Fermi–Löwdin orbital self-interaction correction (FLOSIC) method for practical density functional calculations without SIE.
中文翻译:

Fermi–Löwdin轨道自相互作用校正的密度泛函逼近带来的收缩自相互作用误差
自相互作用误差(SIE)是Kohn-Sham密度泛函理论实际交换相关泛函的主要缺点之一。尽管如此,由于其相对较高的计算成本和对标准现代功能的持续改进,在文献中很少使用从近似密度泛函中明确去除SIE的方法。在本文中,我们评估了Pederson,Ruzsinszky和Perdew [ J. Chem。物理 2014年,140,[121103]用于在基于费米轨道的密度泛函理论中执行无自相互作用的计算。为此,我们采用了由反应能组成的测试装置,这些反应能被认为对SIE特别敏感。我们发现,无参数费米-洛文轨道自相互作用校正方法与标准的局部自旋密度逼近(LSDA)和Perdew-Burke-Ernzerhof(PBE)功能相结合,比其父级LSDA能够更好地估计反应能和PBE官能团可用于这两组反应中的大多数反应。它们的性能也与全局PBE0和范围分隔的LC-ωPBE杂种相当,后者通过包括Hartree-Fock交换而部分消除了SIE。
更新日期:2018-11-09
中文翻译:
Fermi–Löwdin轨道自相互作用校正的密度泛函逼近带来的收缩自相互作用误差
自相互作用误差(SIE)是Kohn-Sham密度泛函理论实际交换相关泛函的主要缺点之一。尽管如此,由于其相对较高的计算成本和对标准现代功能的持续改进,在文献中很少使用从近似密度泛函中明确去除SIE的方法。在本文中,我们评估了Pederson,Ruzsinszky和Perdew [ J. Chem。物理 2014年,140,[121103]用于在基于费米轨道的密度泛函理论中执行无自相互作用的计算。为此,我们采用了由反应能组成的测试装置,这些反应能被认为对SIE特别敏感。我们发现,无参数费米-洛文轨道自相互作用校正方法与标准的局部自旋密度逼近(LSDA)和Perdew-Burke-Ernzerhof(PBE)功能相结合,比其父级LSDA能够更好地估计反应能和PBE官能团可用于这两组反应中的大多数反应。它们的性能也与全局PBE0和范围分隔的LC-ωPBE杂种相当,后者通过包括Hartree-Fock交换而部分消除了SIE。

























 京公网安备 11010802027423号
京公网安备 11010802027423号