当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Multiparameter Optimization of Two Common Proteomics Quantification Methods for Quantifying Low-Abundance Proteins
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2018-11-26 , DOI: 10.1021/acs.jproteome.8b00769 Chengqian Zhang 1 , Zhaomei Shi 1 , Ying Han 1 , Yan Ren 2, 3 , Piliang Hao 1
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2018-11-26 , DOI: 10.1021/acs.jproteome.8b00769 Chengqian Zhang 1 , Zhaomei Shi 1 , Ying Han 1 , Yan Ren 2, 3 , Piliang Hao 1
Affiliation
|
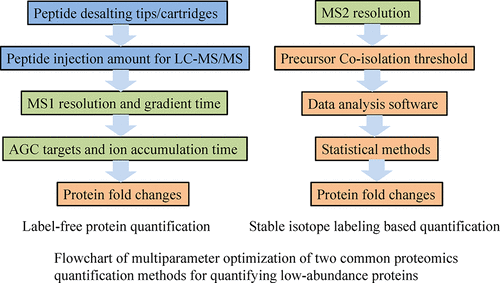
Quantitative proteomics has been extensively applied in the screening of differentially regulated proteins in various research areas for decades, but its sensitivity and accuracy have been a bottleneck for many applications. Every step in the proteomics workflow can potentially affect the quantification of low-abundance proteins, but a systematic evaluation of their effects has not been done yet. In this work, to improve the sensitivity and accuracy of label-free quantification and tandem mass tags (TMT) labeling in quantifying low-abundance proteins, multiparameter optimization was carried out using a complex 2-proteome artificial sample mixture for a series of steps from sample preparation to data analysis, including the desalting of peptides, peptide injection amount for LC-MS/MS, MS1 resolution, the length of LC-MS/MS gradient, AGC targets, ion accumulation time, MS2 resolution, precursor coisolation threshold, data analysis software, statistical calculation methods, and protein fold changes, and the best settings for each parameter were defined. The suitable cutoffs for detecting low-abundance proteins with at least 1.5-fold and 2-fold changes were identified for label-free and TMT methods, respectively. The use of optimized parameters will significantly improve the overall performance of quantitative proteomics in quantifying low-abundance proteins and thus promote its application in other research areas.
中文翻译:

两种常用蛋白质组学定量方法定量低丰度蛋白质的多参数优化
数十年来,定量蛋白质组学已广泛应用于各种研究领域中差异调节蛋白的筛选,但其灵敏度和准确性一直是许多应用的瓶颈。蛋白质组学工作流程的每个步骤都可能影响低丰度蛋白质的定量,但是尚未对其效果进行系统的评估。在这项工作中,为了提高无标记定量和串联质量标签(TMT)标记在定量低丰度蛋白质中的敏感性和准确性,使用复杂的2蛋白质组人工样品混合物进行了多参数优化,从以下步骤开始样品制备以进行数据分析,包括肽脱盐,LC-MS / MS的肽注入量,MS1分辨率,LC-MS / MS梯度的长度,AGC目标 定义了离子累积时间,MS2分辨率,前体共隔离阈值,数据分析软件,统计计算方法和蛋白质倍数变化,并定义了每个参数的最佳设置。对于无标记法和TMT法,分别确定了检测至少有1.5倍和2倍变化的低丰度蛋白质的合适临界值。优化参数的使用将显着提高定量蛋白质组学在定量低丰度蛋白质中的整体性能,从而促进其在其他研究领域中的应用。对于无标记方法和TMT方法,分别鉴定出5倍和2倍的变化。优化参数的使用将显着提高定量蛋白质组学在定量低丰度蛋白质中的整体性能,从而促进其在其他研究领域中的应用。对于无标记方法和TMT方法,分别鉴定出5倍和2倍的变化。优化参数的使用将显着提高定量蛋白质组学在定量低丰度蛋白质中的整体性能,从而促进其在其他研究领域中的应用。
更新日期:2018-11-27
中文翻译:
两种常用蛋白质组学定量方法定量低丰度蛋白质的多参数优化
数十年来,定量蛋白质组学已广泛应用于各种研究领域中差异调节蛋白的筛选,但其灵敏度和准确性一直是许多应用的瓶颈。蛋白质组学工作流程的每个步骤都可能影响低丰度蛋白质的定量,但是尚未对其效果进行系统的评估。在这项工作中,为了提高无标记定量和串联质量标签(TMT)标记在定量低丰度蛋白质中的敏感性和准确性,使用复杂的2蛋白质组人工样品混合物进行了多参数优化,从以下步骤开始样品制备以进行数据分析,包括肽脱盐,LC-MS / MS的肽注入量,MS1分辨率,LC-MS / MS梯度的长度,AGC目标 定义了离子累积时间,MS2分辨率,前体共隔离阈值,数据分析软件,统计计算方法和蛋白质倍数变化,并定义了每个参数的最佳设置。对于无标记法和TMT法,分别确定了检测至少有1.5倍和2倍变化的低丰度蛋白质的合适临界值。优化参数的使用将显着提高定量蛋白质组学在定量低丰度蛋白质中的整体性能,从而促进其在其他研究领域中的应用。对于无标记方法和TMT方法,分别鉴定出5倍和2倍的变化。优化参数的使用将显着提高定量蛋白质组学在定量低丰度蛋白质中的整体性能,从而促进其在其他研究领域中的应用。对于无标记方法和TMT方法,分别鉴定出5倍和2倍的变化。优化参数的使用将显着提高定量蛋白质组学在定量低丰度蛋白质中的整体性能,从而促进其在其他研究领域中的应用。

























 京公网安备 11010802027423号
京公网安备 11010802027423号