当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Electronic processes in NO dimerization on Ag and Cu clusters: DFT and MRMP2 studies
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-10-31 , DOI: 10.1002/jcc.25568 Nozomi Takagi 1 , Masayuki Nakagaki 2 , Kazuya Ishimura 3 , Ryoichi Fukuda 1 , Masahiro Ehara 1, 3 , Shigeyoshi Sakaki 1, 2
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-10-31 , DOI: 10.1002/jcc.25568 Nozomi Takagi 1 , Masayuki Nakagaki 2 , Kazuya Ishimura 3 , Ryoichi Fukuda 1 , Masahiro Ehara 1, 3 , Shigeyoshi Sakaki 1, 2
Affiliation
|
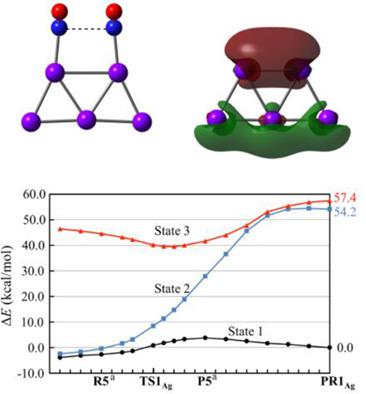
Experimentally observed NO dimerization on Cu and Ag surfaces is surprising because binding energy of NO dimer is very small in gas phase. MRMP2, MP2 to MP4, CCSD(T), and DFT studies of NO dimerization on Ag2 and Cu2 clusters disclosed that the CCSD(T) method could be applied to this reaction on Ag2 and Cu2 unlike NO dimerization in gas phase which exhibits significantly large nondynamical electron correlation effect. Charge‐transfer (CT) from Ag2 and Cu2 to NO moieties plays important role in NN bond formation between two NO molecules. This CT considerably decreases nondynamical correlation effect. Also, the DFT method could be applied to this NO dimerization, if appropriate DFT functional is used; all pure functionals examined here and most of the hybrid functionals underestimated the activation barrier (Ea), while only ωB97X provided Ea similar to CCSD(T)‐calculated value. NO dimerization on similar Cu2 and Cu5 needs moderately larger Ea than those on Ag2 and Ag5, because frontier orbital participating in the CT exists at lower energy in Cu2 and Cu5 than in Ag2 and Ag5. The Ea decreases in the order Ag2 >> Ag38 > Ag7 ∼ Ag5 and the reaction energy (ΔE) is positive (endothermic) in Ag2 but significantly negative in Ag38, Ag7, and Ag5, indicating that various Ag clusters could be effective for NO dimerization except for Ag2. The decreasing order of Ea and increasing order of exothermicity are attributed to increasing order of the frontier orbital energy of Ag2 < Ag38 < Ag7 ∼ Ag5. © 2018 Wiley Periodicals, Inc.
中文翻译:

Ag 和 Cu 簇上 NO 二聚化的电子过程:DFT 和 MRMP2 研究
实验观察到的 Cu 和 Ag 表面的 NO 二聚化是令人惊讶的,因为 NO 二聚体在气相中的结合能非常小。MRMP2、MP2 到 MP4、CCSD(T) 和 DFT 对 Ag2 和 Cu2 簇上 NO 二聚化的研究表明,CCSD(T) 方法可以应用于这种在 Ag2 和 Cu2 上的反应,不像气相中的 NO 二聚化表现出明显的大非动态电子相关效应。从 Ag2 和 Cu2 到 NO 部分的电荷转移 (CT) 在两个 NO 分子之间形成 NN 键中起重要作用。该 CT 显着降低了非动力相关效应。此外,如果使用适当的 DFT 泛函,DFT 方法可以应用于这种 NO 二聚化;这里检查的所有纯泛函和大多数混合泛函都低估了激活屏障 (Ea),而只有 ωB97X 提供类似于 CCSD(T) 计算值的 Ea。在类似的 Cu2 和 Cu5 上的 NO 二聚化需要比在 Ag2 和 Ag5 上的那些稍大的 Ea,因为参与 CT 的前沿轨道在 Cu2 和 Cu5 中存在的能量低于 Ag2 和 Ag5。Ea 以 Ag2 >> Ag38 > Ag7 ∼ Ag5 的顺序降低,Ag2 中的反应能 (ΔE) 为正(吸热),而 Ag38、Ag7 和 Ag5 中的反应能(ΔE)显着为负,表明各种 Ag 簇对 NO 二聚化有效除了 Ag2。Ea 的递减顺序和放热性的递增顺序归因于 Ag2 < Ag38 < Ag7 ∼ Ag5 的前线轨道能量递增顺序。© 2018 Wiley Periodicals, Inc. 因为参与 CT 的前沿轨道在 Cu2 和 Cu5 中的能量低于 Ag2 和 Ag5。Ea 以 Ag2 >> Ag38 > Ag7 ∼ Ag5 的顺序降低,Ag2 中的反应能 (ΔE) 为正(吸热),而 Ag38、Ag7 和 Ag5 中的反应能(ΔE)显着为负,表明各种 Ag 簇对 NO 二聚化有效除了 Ag2。Ea 的递减顺序和放热性的递增顺序归因于 Ag2 < Ag38 < Ag7 ∼ Ag5 的前线轨道能量递增顺序。© 2018 Wiley Periodicals, Inc. 因为参与 CT 的前沿轨道在 Cu2 和 Cu5 中的能量低于 Ag2 和 Ag5。Ea 以 Ag2 >> Ag38 > Ag7 ∼ Ag5 的顺序降低,Ag2 中的反应能 (ΔE) 为正(吸热),而 Ag38、Ag7 和 Ag5 中的反应能(ΔE)显着为负,表明各种 Ag 簇对 NO 二聚化有效除了 Ag2。Ea 的递减顺序和放热性的递增顺序归因于 Ag2 < Ag38 < Ag7 ∼ Ag5 的前线轨道能量递增顺序。© 2018 Wiley Periodicals, Inc. 表明除 Ag2 外,各种 Ag 簇对 NO 二聚化均有效。Ea 的递减顺序和放热性的递增顺序归因于 Ag2 < Ag38 < Ag7 ∼ Ag5 的前线轨道能量递增顺序。© 2018 Wiley Periodicals, Inc. 表明除 Ag2 外,各种 Ag 簇对 NO 二聚化均有效。Ea 的递减顺序和放热性的递增顺序归因于 Ag2 < Ag38 < Ag7 ∼ Ag5 的前线轨道能量递增顺序。© 2018 Wiley Periodicals, Inc.
更新日期:2018-10-31
中文翻译:
Ag 和 Cu 簇上 NO 二聚化的电子过程:DFT 和 MRMP2 研究
实验观察到的 Cu 和 Ag 表面的 NO 二聚化是令人惊讶的,因为 NO 二聚体在气相中的结合能非常小。MRMP2、MP2 到 MP4、CCSD(T) 和 DFT 对 Ag2 和 Cu2 簇上 NO 二聚化的研究表明,CCSD(T) 方法可以应用于这种在 Ag2 和 Cu2 上的反应,不像气相中的 NO 二聚化表现出明显的大非动态电子相关效应。从 Ag2 和 Cu2 到 NO 部分的电荷转移 (CT) 在两个 NO 分子之间形成 NN 键中起重要作用。该 CT 显着降低了非动力相关效应。此外,如果使用适当的 DFT 泛函,DFT 方法可以应用于这种 NO 二聚化;这里检查的所有纯泛函和大多数混合泛函都低估了激活屏障 (Ea),而只有 ωB97X 提供类似于 CCSD(T) 计算值的 Ea。在类似的 Cu2 和 Cu5 上的 NO 二聚化需要比在 Ag2 和 Ag5 上的那些稍大的 Ea,因为参与 CT 的前沿轨道在 Cu2 和 Cu5 中存在的能量低于 Ag2 和 Ag5。Ea 以 Ag2 >> Ag38 > Ag7 ∼ Ag5 的顺序降低,Ag2 中的反应能 (ΔE) 为正(吸热),而 Ag38、Ag7 和 Ag5 中的反应能(ΔE)显着为负,表明各种 Ag 簇对 NO 二聚化有效除了 Ag2。Ea 的递减顺序和放热性的递增顺序归因于 Ag2 < Ag38 < Ag7 ∼ Ag5 的前线轨道能量递增顺序。© 2018 Wiley Periodicals, Inc. 因为参与 CT 的前沿轨道在 Cu2 和 Cu5 中的能量低于 Ag2 和 Ag5。Ea 以 Ag2 >> Ag38 > Ag7 ∼ Ag5 的顺序降低,Ag2 中的反应能 (ΔE) 为正(吸热),而 Ag38、Ag7 和 Ag5 中的反应能(ΔE)显着为负,表明各种 Ag 簇对 NO 二聚化有效除了 Ag2。Ea 的递减顺序和放热性的递增顺序归因于 Ag2 < Ag38 < Ag7 ∼ Ag5 的前线轨道能量递增顺序。© 2018 Wiley Periodicals, Inc. 因为参与 CT 的前沿轨道在 Cu2 和 Cu5 中的能量低于 Ag2 和 Ag5。Ea 以 Ag2 >> Ag38 > Ag7 ∼ Ag5 的顺序降低,Ag2 中的反应能 (ΔE) 为正(吸热),而 Ag38、Ag7 和 Ag5 中的反应能(ΔE)显着为负,表明各种 Ag 簇对 NO 二聚化有效除了 Ag2。Ea 的递减顺序和放热性的递增顺序归因于 Ag2 < Ag38 < Ag7 ∼ Ag5 的前线轨道能量递增顺序。© 2018 Wiley Periodicals, Inc. 表明除 Ag2 外,各种 Ag 簇对 NO 二聚化均有效。Ea 的递减顺序和放热性的递增顺序归因于 Ag2 < Ag38 < Ag7 ∼ Ag5 的前线轨道能量递增顺序。© 2018 Wiley Periodicals, Inc. 表明除 Ag2 外,各种 Ag 簇对 NO 二聚化均有效。Ea 的递减顺序和放热性的递增顺序归因于 Ag2 < Ag38 < Ag7 ∼ Ag5 的前线轨道能量递增顺序。© 2018 Wiley Periodicals, Inc.

























 京公网安备 11010802027423号
京公网安备 11010802027423号