当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Experimental Evidence of Synergistic Interactions in Pyrrole–Phenol Complexes at Low Temperatures under Isolated Conditions
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2018-10-30 00:00:00 , DOI: 10.1021/acs.jpca.8b09076 Shubhra Sarkar 1 , N. Ramanathan 1 , K. Sundararajan 1
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2018-10-30 00:00:00 , DOI: 10.1021/acs.jpca.8b09076 Shubhra Sarkar 1 , N. Ramanathan 1 , K. Sundararajan 1
Affiliation
|
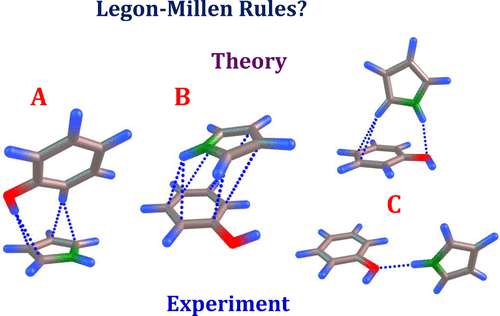
The simultaneous possession of π-electron clouds and acidic hydrogen atoms in pyrrole (C4H5N) and phenol (C6H5OH) framework opens the potentiality in exploring the synergistic interactions in their weakly bonded complexes. In this work, the synergistic hydrogen bonding in C4H5N–C6H5OH complexes is therefore investigated using FTIR spectroscopy under isolated conditions at low temperatures. Computations performed at DFT, DFT-GD3, M06, and MP2 level of theories employing aug-cc-pVDZ basis set yielded three minima on the potential energy surface for the 1:1 complex of C4H5N–C6H5OH. All three optimized structures showed synergistic interactions, where both C6H5OH and C4H5N simultaneously act as a proton donor and acceptor at MP2/aug-cc-pVDZ level of theory. In the global minimum complex A, the hydroxyl proton and the C–H group of C6H5OH interact with the π-cloud of C4H5N. The first local minimum corresponds to complex B, where the N–H and π-electrons of C4H5N interact with π-electrons of C6H5OH. In complex C, the N–H and C–H groups of C4H5N interact with O–H and π-cloud of C6H5OH, respectively. Complex A was the lowest energy structure at all levels of theory, whereas the stabilization energies of complexes B and C varied depending upon the levels of theory used. Interestingly, the stabilization energies as predicted by the DFT method are in accordance with Etter’s and Legon–Millen rules; however, a deviation in the Legon–Millen rule was discerned with empirical (DFT-GD3, M06) and dispersion corrected (MP2) methods. On comparing the experimental vibrational wavenumber shifts in the N–H stretching and bending modes of C4H5N and O–H stretching mode of C6H5OH submolecules with the computed shifts, all three complexes were identified in the N2 matrix. Natural Bond Orbital and Energy Decomposition analyses were performed to characterize the nature of the synergistic interaction in these complexes.
中文翻译:

隔离条件下低温下吡咯-苯酚配合物协同相互作用的实验证据
在吡咯(C 4 H 5 N)和苯酚(C 6 H 5 OH)骨架中同时拥有π电子云和酸性氢原子为探索其弱键合配合物的协同相互作用提供了潜力。因此,在这项工作中,我们使用FTIR光谱技术在低温下在孤立条件下研究了C 4 H 5 N–C 6 H 5 OH配合物中的协同氢键。在DFT,DFT-GD3,M06和MP2级别上使用aug-cc-pVDZ基集进行的计算在C 4 H 5 N–C 1:1络合物的势能面上产生了三个极小值6 H 5 OH。所有这三个优化的结构均显示出协同相互作用,其中C 6 H 5 OH和C 4 H 5 N同时在理论上为MP2 / aug-cc-pVDZ充当质子供体和受体。在全局最小络合物A中,C 6 H 5 OH的羟基质子和C–H基团与C 4 H 5 N的π云相互作用。第一个局部最小值对应于络合物B,其中N–H和C 4 H 5 N的π电子与C 6 H 5 OH的π电子相互作用。在复数C中,C 4的N–H和C–H组H 5 N分别与C 6 H 5 OH的OH和π云相互作用。络合物A在所有理论水平上都是最低的能量结构,而络合物B和C的稳定能根据所用理论水平而变化。有趣的是,通过DFT方法预测的稳定能符合Etter和Legon-Millen的规则。但是,经验法(DFT-GD3,M06)和色散校正法(MP2)可以识别出Legon-Millen规则的偏差。上比较在N-H实验振动波数位移拉伸和弯曲的C模式4 ħ 5 N和O-H伸缩的C模式6 ħ 5具有计算位移的OH亚分子,在N 2基质中鉴定了所有三种配合物。进行了自然键轨道和能量分解分析,以表征这些复合物中协同相互作用的性质。
更新日期:2018-10-30
中文翻译:
隔离条件下低温下吡咯-苯酚配合物协同相互作用的实验证据
在吡咯(C 4 H 5 N)和苯酚(C 6 H 5 OH)骨架中同时拥有π电子云和酸性氢原子为探索其弱键合配合物的协同相互作用提供了潜力。因此,在这项工作中,我们使用FTIR光谱技术在低温下在孤立条件下研究了C 4 H 5 N–C 6 H 5 OH配合物中的协同氢键。在DFT,DFT-GD3,M06和MP2级别上使用aug-cc-pVDZ基集进行的计算在C 4 H 5 N–C 1:1络合物的势能面上产生了三个极小值6 H 5 OH。所有这三个优化的结构均显示出协同相互作用,其中C 6 H 5 OH和C 4 H 5 N同时在理论上为MP2 / aug-cc-pVDZ充当质子供体和受体。在全局最小络合物A中,C 6 H 5 OH的羟基质子和C–H基团与C 4 H 5 N的π云相互作用。第一个局部最小值对应于络合物B,其中N–H和C 4 H 5 N的π电子与C 6 H 5 OH的π电子相互作用。在复数C中,C 4的N–H和C–H组H 5 N分别与C 6 H 5 OH的OH和π云相互作用。络合物A在所有理论水平上都是最低的能量结构,而络合物B和C的稳定能根据所用理论水平而变化。有趣的是,通过DFT方法预测的稳定能符合Etter和Legon-Millen的规则。但是,经验法(DFT-GD3,M06)和色散校正法(MP2)可以识别出Legon-Millen规则的偏差。上比较在N-H实验振动波数位移拉伸和弯曲的C模式4 ħ 5 N和O-H伸缩的C模式6 ħ 5具有计算位移的OH亚分子,在N 2基质中鉴定了所有三种配合物。进行了自然键轨道和能量分解分析,以表征这些复合物中协同相互作用的性质。


























 京公网安备 11010802027423号
京公网安备 11010802027423号