当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Can large active-space CASSCF calculation make sense to the reaction analysis of iron complex? A benchmark study of methane oxidation reaction by FeO+
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-10-23 , DOI: 10.1002/jcc.25640 Naoki Nakatani 1 , Masahiko Hada 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-10-23 , DOI: 10.1002/jcc.25640 Naoki Nakatani 1 , Masahiko Hada 1
Affiliation
|
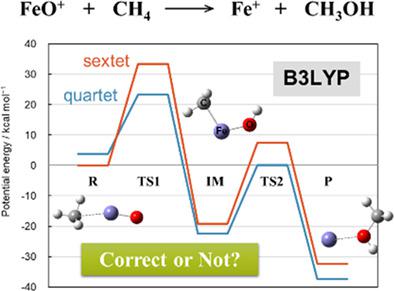
A methane oxidation reaction by FeO+ cation was theoretically investigated based on the density functional theory (DFT) and the complete active‐space self‐consistent field (CASSCF) method as well as the coupled‐cluster singles, doubles, and perturbative triples (CCSD(T)) to explore the active‐space dependency to computational analyses in such strongly correlated reaction systems. A small active‐space CASSCF(5e in 5o) calculation, which only includes five 3d orbitals of the Fe atom in the active‐space, showed remarkable difference both in energy and geometry compared to those computed by the DFT and CCSD(T) methods. Interestingly, a large active‐space CASSCF(17e in 17o) calculation, which includes almost all the valence orbitals gives a qualitative agreement with either the DFT or the CCSD(T) results in the first half part of the reaction, although it varies from them in the latter half part. Therefore, it is indicated that the active‐space dependency is serious in some part of the reaction and the small active‐space CASSCF might lead a wrong discussion. We further investigated the optimized geometry of the intermediate complex with the small and the large active‐space CASSCF methods as well as the CCSD(T) method, and found that the CASSCF(5e in 5o)‐optimized geometry is considerably different from the others. In consequence, a small active‐space CASSCF/CASPT2 calculation does not really work for such a strongly correlated reaction system even qualitatively, and a sophisticated assessment using the large active‐space CASSCF/CASPT2 method will be indispensable. © 2018 Wiley Periodicals, Inc.
中文翻译:

大活性空间CASSCF计算对铁配合物的反应分析有意义吗?FeO+对甲烷氧化反应的基准研究
基于密度泛函理论(DFT)和完全活性空间自洽场(CASSCF)方法以及耦合簇单峰、双峰和微扰三元组(CCSD( T)) 在这种强相关反应系统中探索活性空间对计算分析的依赖性。一个小的活性空间 CASSCF(5e in 5o) 计算,只包括活性空间中 Fe 原子的 5 个 3d 轨道,与 DFT 和 CCSD(T) 方法计算的结果相比,在能量和几何结构上都有显着差异. 有趣的是,包含几乎所有价轨道的大型活性空间 CASSCF(17e in 17o) 计算在反应的前半部分与 DFT 或 CCSD(T) 结果定性一致,虽然后半部分与他们有所不同。因此,这表明反应的某些部分的活性空间依赖性很严重,小的活性空间CASSCF可能会导致错误的讨论。我们使用小型和大型活性空间 CASSCF 方法以及 CCSD(T) 方法进一步研究了中间复合体的优化几何形状,发现 CASSCF(5e in 5o) 优化几何形状与其他几何形状有很大不同. 因此,即使在定性上,小的活性空间 CASSCF/CASPT2 计算也不适用于如此强相关的反应系统,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc. 这表明反应的某些部分的活性空间依赖性很严重,小的活性空间CASSCF可能会导致错误的讨论。我们使用小型和大型活性空间 CASSCF 方法以及 CCSD(T) 方法进一步研究了中间复合体的优化几何形状,发现 CASSCF(5e in 5o) 优化几何形状与其他几何形状有很大不同. 因此,即使在定性上,小的活性空间 CASSCF/CASPT2 计算也不适用于如此强相关的反应系统,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc. 这表明反应的某些部分的活性空间依赖性很严重,小的活性空间CASSCF可能会导致错误的讨论。我们使用小型和大型活性空间 CASSCF 方法以及 CCSD(T) 方法进一步研究了中间复合体的优化几何形状,发现 CASSCF(5e in 5o) 优化几何形状与其他几何形状有很大不同. 因此,即使在定性上,小的活性空间 CASSCF/CASPT2 计算也不适用于如此强相关的反应系统,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc. 我们使用小型和大型活性空间 CASSCF 方法以及 CCSD(T) 方法进一步研究了中间复合体的优化几何形状,发现 CASSCF(5e in 5o) 优化几何形状与其他几何形状有很大不同. 因此,即使在定性上,小的活性空间 CASSCF/CASPT2 计算也不适用于如此强相关的反应系统,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc. 我们使用小型和大型活性空间 CASSCF 方法以及 CCSD(T) 方法进一步研究了中间复合体的优化几何形状,发现 CASSCF(5e in 5o) 优化几何形状与其他几何形状有很大不同. 因此,即使在定性上,小的活性空间 CASSCF/CASPT2 计算也不适用于如此强相关的反应系统,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc. 一个小的活性空间 CASSCF/CASPT2 计算对于如此强相关的反应系统甚至在定性上都不起作用,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc. 一个小的活性空间 CASSCF/CASPT2 计算对于如此强相关的反应系统甚至在定性上都不起作用,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc.
更新日期:2018-10-23
中文翻译:
大活性空间CASSCF计算对铁配合物的反应分析有意义吗?FeO+对甲烷氧化反应的基准研究
基于密度泛函理论(DFT)和完全活性空间自洽场(CASSCF)方法以及耦合簇单峰、双峰和微扰三元组(CCSD( T)) 在这种强相关反应系统中探索活性空间对计算分析的依赖性。一个小的活性空间 CASSCF(5e in 5o) 计算,只包括活性空间中 Fe 原子的 5 个 3d 轨道,与 DFT 和 CCSD(T) 方法计算的结果相比,在能量和几何结构上都有显着差异. 有趣的是,包含几乎所有价轨道的大型活性空间 CASSCF(17e in 17o) 计算在反应的前半部分与 DFT 或 CCSD(T) 结果定性一致,虽然后半部分与他们有所不同。因此,这表明反应的某些部分的活性空间依赖性很严重,小的活性空间CASSCF可能会导致错误的讨论。我们使用小型和大型活性空间 CASSCF 方法以及 CCSD(T) 方法进一步研究了中间复合体的优化几何形状,发现 CASSCF(5e in 5o) 优化几何形状与其他几何形状有很大不同. 因此,即使在定性上,小的活性空间 CASSCF/CASPT2 计算也不适用于如此强相关的反应系统,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc. 这表明反应的某些部分的活性空间依赖性很严重,小的活性空间CASSCF可能会导致错误的讨论。我们使用小型和大型活性空间 CASSCF 方法以及 CCSD(T) 方法进一步研究了中间复合体的优化几何形状,发现 CASSCF(5e in 5o) 优化几何形状与其他几何形状有很大不同. 因此,即使在定性上,小的活性空间 CASSCF/CASPT2 计算也不适用于如此强相关的反应系统,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc. 这表明反应的某些部分的活性空间依赖性很严重,小的活性空间CASSCF可能会导致错误的讨论。我们使用小型和大型活性空间 CASSCF 方法以及 CCSD(T) 方法进一步研究了中间复合体的优化几何形状,发现 CASSCF(5e in 5o) 优化几何形状与其他几何形状有很大不同. 因此,即使在定性上,小的活性空间 CASSCF/CASPT2 计算也不适用于如此强相关的反应系统,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc. 我们使用小型和大型活性空间 CASSCF 方法以及 CCSD(T) 方法进一步研究了中间复合体的优化几何形状,发现 CASSCF(5e in 5o) 优化几何形状与其他几何形状有很大不同. 因此,即使在定性上,小的活性空间 CASSCF/CASPT2 计算也不适用于如此强相关的反应系统,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc. 我们使用小型和大型活性空间 CASSCF 方法以及 CCSD(T) 方法进一步研究了中间复合体的优化几何形状,发现 CASSCF(5e in 5o) 优化几何形状与其他几何形状有很大不同. 因此,即使在定性上,小的活性空间 CASSCF/CASPT2 计算也不适用于如此强相关的反应系统,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc. 一个小的活性空间 CASSCF/CASPT2 计算对于如此强相关的反应系统甚至在定性上都不起作用,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc. 一个小的活性空间 CASSCF/CASPT2 计算对于如此强相关的反应系统甚至在定性上都不起作用,使用大活性空间 CASSCF/CASPT2 方法进行复杂的评估将是必不可少的。© 2018 Wiley Periodicals, Inc.


























 京公网安备 11010802027423号
京公网安备 11010802027423号