当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
MDockPeP: An ab-initio protein-peptide docking server
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-10-23 , DOI: 10.1002/jcc.25555 Xianjin Xu 1 , Chengfei Yan 1 , Xiaoqin Zou 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-10-23 , DOI: 10.1002/jcc.25555 Xianjin Xu 1 , Chengfei Yan 1 , Xiaoqin Zou 1
Affiliation
|
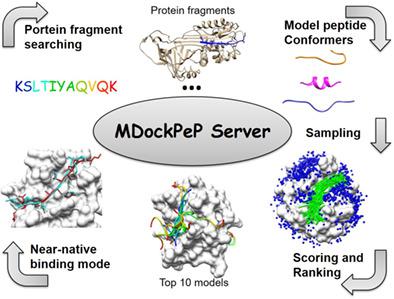
Protein–peptide interactions play a crucial role in a variety of cellular processes. The protein–peptide complex structure is a key to understand the mechanisms underlying protein–peptide interactions and is critical for peptide therapeutic development. We present a user‐friendly protein–peptide docking server, MDockPeP. Starting from a peptide sequence and a protein receptor structure, the MDockPeP Server globally docks the all‐atom, flexible peptide to the protein receptor. The produced modes are then evaluated with a statistical potential‐based scoring function, ITScorePeP. This method was systematically validated using the peptiDB benchmarking database. At least one near‐native peptide binding mode was ranked among top 10 (or top 500) in 59% (85%) of the bound cases, and in 40.6% (71.9%) of the challenging unbound cases. The server can be used for both protein–peptide complex structure prediction and initial‐stage sampling of the protein–peptide binding modes for other docking or simulation methods. MDockPeP Server is freely available at http://zougrouptoolkit.missouri.edu/mdockpep. © 2018 Wiley Periodicals, Inc.
中文翻译:

MDockPeP:一个从头开始的蛋白质肽对接服务器
蛋白质-肽相互作用在各种细胞过程中起着至关重要的作用。蛋白质-肽复合结构是了解蛋白质-肽相互作用机制的关键,对肽治疗开发至关重要。我们提出了一个用户友好的蛋白质肽对接服务器,MDockPeP。从肽序列和蛋白质受体结构开始,MDockPeP 服务器在全局范围内将全原子、灵活的肽与蛋白质受体对接。然后使用基于统计潜力的评分函数 ITScorePeP 评估产生的模式。该方法使用 peptiDB 基准数据库进行了系统验证。在 59% (85%) 的绑定案例和 40.6% (71.9%) 的具有挑战性的未绑定案例中,至少有一种近天然肽结合模式进入前 10 名(或前 500 名)。该服务器可用于其他对接或模拟方法的蛋白质 - 肽复合结构预测和蛋白质 - 肽结合模式的初始阶段采样。MDockPeP 服务器可在 http://zougrouptoolkit.missouri.edu/mdockpep 免费获得。© 2018 Wiley Periodicals, Inc.
更新日期:2018-10-23
中文翻译:
MDockPeP:一个从头开始的蛋白质肽对接服务器
蛋白质-肽相互作用在各种细胞过程中起着至关重要的作用。蛋白质-肽复合结构是了解蛋白质-肽相互作用机制的关键,对肽治疗开发至关重要。我们提出了一个用户友好的蛋白质肽对接服务器,MDockPeP。从肽序列和蛋白质受体结构开始,MDockPeP 服务器在全局范围内将全原子、灵活的肽与蛋白质受体对接。然后使用基于统计潜力的评分函数 ITScorePeP 评估产生的模式。该方法使用 peptiDB 基准数据库进行了系统验证。在 59% (85%) 的绑定案例和 40.6% (71.9%) 的具有挑战性的未绑定案例中,至少有一种近天然肽结合模式进入前 10 名(或前 500 名)。该服务器可用于其他对接或模拟方法的蛋白质 - 肽复合结构预测和蛋白质 - 肽结合模式的初始阶段采样。MDockPeP 服务器可在 http://zougrouptoolkit.missouri.edu/mdockpep 免费获得。© 2018 Wiley Periodicals, Inc.


























 京公网安备 11010802027423号
京公网安备 11010802027423号