当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
FALDI-based criterion for and the origin of an electron density bridge with an associated (3,-1) critical point on Bader's molecular graph
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-10-14 , DOI: 10.1002/jcc.25548 Jurgens H. de Lange 1 , Daniël M. E. van Niekerk 1 , Ignacy Cukrowski 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-10-14 , DOI: 10.1002/jcc.25548 Jurgens H. de Lange 1 , Daniël M. E. van Niekerk 1 , Ignacy Cukrowski 1
Affiliation
|
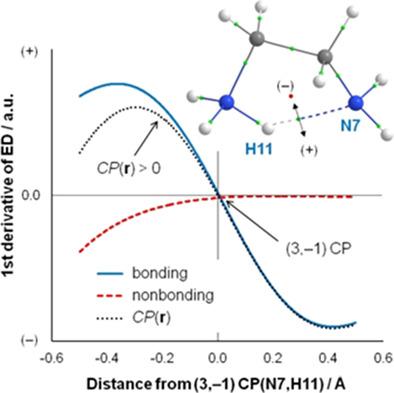
The total electron density (ED) along the λ2‐eigenvector is decomposed into contributions which either facilitate or hinder the presence of an electron density bridge (DB, often called an atomic interaction line or a bond path). Our FALDI‐based approach explains a DB presence as a result of a dominating rate of change of facilitating factors relative to the rate of change of hindering factors; a novel and universal criterion for a DB presence is, thus, proposed. Importantly, facilitating factors show, in absolute terms, a concentration of ED in the internuclear region as commonly observed for most chemical bonds, whereas hindering factors show a depletion of ED in the internuclear region. We test our approach on four intramolecular interactions, namely (i) an attractive classical H‐bond, (ii) a repulsive O⋅⋅⋅O interaction, (iii) an attractive Cl⋅⋅⋅Cl interaction, and (iv) an attractive CH⋅⋅⋅HC interaction. (Dis)appearance of a DB is (i) shown to be due to a “small” change in molecular environment and (ii) qualitatively and quantitatively linked with specific atoms and atom‐pairs. The protocol described is equally applicable (a) to any internuclear region, (b) regardless of what kind of interaction (attractive/repulsive) atoms are involved in, (c) at any level of theory used to compute the molecular structure and corresponding wavefunction, and (d) equilibrium or nonequilibrium structures. Finally, we argue for a paradigm shift in the description of chemical interactions, from the ED perspective, in favor of a multicenter rather than diatomic approach in interpreting ED distributions in internuclear regions. © 2018 Wiley Periodicals, Inc.
中文翻译:

基于 FALDI 的电子密度桥的标准和起源,在 Bader 的分子图上有一个相关的 (3,-1) 临界点
沿 λ2 特征向量的总电子密度 (ED) 被分解为促进或阻碍电子密度桥(DB,通常称为原子相互作用线或键路径)存在的贡献。我们基于 FALDI 的方法将 DB 的存在解释为促进因素相对于阻碍因素的变化率占主导地位的变化率;因此,提出了一种新的、通用的数据库存在标准。重要的是,从绝对值来看,促进因素显示了核间区域中 ED 的浓度,正如大多数化学键通常观察到的那样,而阻碍因素显示核间区域中 ED 的消耗。我们在四种分子内相互作用上测试我们的方法,即(i)有吸引力的经典氢键,(ii)排斥性 O⋅⋅⋅O 相互作用,(iii) 有吸引力的 Cl⋅⋅⋅Cl 相互作用,以及 (iv) 有吸引力的 CH⋅⋅⋅HC 相互作用。DB 的(消失)出现是(i)显示是由于分子环境的“小”变化和(ii)与特定原子和原子对定性和定量相关。所描述的协议同样适用于 (a) 任何核间区域, (b) 无论涉及哪种相互作用 (吸引力/排斥) 原子, (c) 在用于计算分子结构和相应波函数的任何理论水平, 和 (d) 平衡或非平衡结构。最后,我们主张从 ED 的角度对化学相互作用的描述进行范式转变,以支持多中心而不是双原子方法来解释核间区域的 ED 分布。© 2018 Wiley Periodicals, Inc. (iv) 有吸引力的 CH⋅⋅⋅HC 相互作用。DB 的 (Dis) 外观 (i) 显示是由于分子环境的“小”变化和 (ii) 与特定原子和原子对定性和定量相关。所描述的协议同样适用于 (a) 任何核间区域, (b) 无论涉及哪种相互作用 (吸引力/排斥) 原子, (c) 在用于计算分子结构和相应波函数的任何理论水平, 和 (d) 平衡或非平衡结构。最后,我们主张从 ED 的角度对化学相互作用的描述进行范式转变,以支持多中心而不是双原子方法来解释核间区域的 ED 分布。© 2018 Wiley Periodicals, Inc. (iv) 有吸引力的 CH⋅⋅⋅HC 相互作用。DB 的(消失)出现是(i)显示是由于分子环境的“小”变化和(ii)与特定原子和原子对定性和定量相关。所描述的协议同样适用于 (a) 任何核间区域, (b) 无论涉及哪种相互作用 (吸引力/排斥) 原子, (c) 在用于计算分子结构和相应波函数的任何理论水平, 和 (d) 平衡或非平衡结构。最后,我们主张从 ED 的角度对化学相互作用的描述进行范式转变,以支持多中心而不是双原子方法来解释核间区域的 ED 分布。© 2018 Wiley Periodicals, Inc. DB 的(消失)出现是(i)显示是由于分子环境的“小”变化和(ii)与特定原子和原子对定性和定量相关。所描述的协议同样适用于 (a) 任何核间区域, (b) 无论涉及哪种相互作用 (吸引力/排斥) 原子, (c) 在用于计算分子结构和相应波函数的任何理论水平, 和 (d) 平衡或非平衡结构。最后,我们主张从 ED 的角度对化学相互作用的描述进行范式转变,以支持多中心而不是双原子方法来解释核间区域的 ED 分布。© 2018 Wiley Periodicals, Inc. DB 的(消失)出现是(i)显示是由于分子环境的“小”变化和(ii)与特定原子和原子对定性和定量相关。所描述的协议同样适用于 (a) 任何核间区域, (b) 无论涉及哪种相互作用 (吸引力/排斥) 原子, (c) 在用于计算分子结构和相应波函数的任何理论水平, 和 (d) 平衡或非平衡结构。最后,我们主张从 ED 的角度对化学相互作用的描述进行范式转变,以支持多中心而不是双原子方法来解释核间区域的 ED 分布。© 2018 Wiley Periodicals, Inc.
更新日期:2018-10-14
中文翻译:
基于 FALDI 的电子密度桥的标准和起源,在 Bader 的分子图上有一个相关的 (3,-1) 临界点
沿 λ2 特征向量的总电子密度 (ED) 被分解为促进或阻碍电子密度桥(DB,通常称为原子相互作用线或键路径)存在的贡献。我们基于 FALDI 的方法将 DB 的存在解释为促进因素相对于阻碍因素的变化率占主导地位的变化率;因此,提出了一种新的、通用的数据库存在标准。重要的是,从绝对值来看,促进因素显示了核间区域中 ED 的浓度,正如大多数化学键通常观察到的那样,而阻碍因素显示核间区域中 ED 的消耗。我们在四种分子内相互作用上测试我们的方法,即(i)有吸引力的经典氢键,(ii)排斥性 O⋅⋅⋅O 相互作用,(iii) 有吸引力的 Cl⋅⋅⋅Cl 相互作用,以及 (iv) 有吸引力的 CH⋅⋅⋅HC 相互作用。DB 的(消失)出现是(i)显示是由于分子环境的“小”变化和(ii)与特定原子和原子对定性和定量相关。所描述的协议同样适用于 (a) 任何核间区域, (b) 无论涉及哪种相互作用 (吸引力/排斥) 原子, (c) 在用于计算分子结构和相应波函数的任何理论水平, 和 (d) 平衡或非平衡结构。最后,我们主张从 ED 的角度对化学相互作用的描述进行范式转变,以支持多中心而不是双原子方法来解释核间区域的 ED 分布。© 2018 Wiley Periodicals, Inc. (iv) 有吸引力的 CH⋅⋅⋅HC 相互作用。DB 的 (Dis) 外观 (i) 显示是由于分子环境的“小”变化和 (ii) 与特定原子和原子对定性和定量相关。所描述的协议同样适用于 (a) 任何核间区域, (b) 无论涉及哪种相互作用 (吸引力/排斥) 原子, (c) 在用于计算分子结构和相应波函数的任何理论水平, 和 (d) 平衡或非平衡结构。最后,我们主张从 ED 的角度对化学相互作用的描述进行范式转变,以支持多中心而不是双原子方法来解释核间区域的 ED 分布。© 2018 Wiley Periodicals, Inc. (iv) 有吸引力的 CH⋅⋅⋅HC 相互作用。DB 的(消失)出现是(i)显示是由于分子环境的“小”变化和(ii)与特定原子和原子对定性和定量相关。所描述的协议同样适用于 (a) 任何核间区域, (b) 无论涉及哪种相互作用 (吸引力/排斥) 原子, (c) 在用于计算分子结构和相应波函数的任何理论水平, 和 (d) 平衡或非平衡结构。最后,我们主张从 ED 的角度对化学相互作用的描述进行范式转变,以支持多中心而不是双原子方法来解释核间区域的 ED 分布。© 2018 Wiley Periodicals, Inc. DB 的(消失)出现是(i)显示是由于分子环境的“小”变化和(ii)与特定原子和原子对定性和定量相关。所描述的协议同样适用于 (a) 任何核间区域, (b) 无论涉及哪种相互作用 (吸引力/排斥) 原子, (c) 在用于计算分子结构和相应波函数的任何理论水平, 和 (d) 平衡或非平衡结构。最后,我们主张从 ED 的角度对化学相互作用的描述进行范式转变,以支持多中心而不是双原子方法来解释核间区域的 ED 分布。© 2018 Wiley Periodicals, Inc. DB 的(消失)出现是(i)显示是由于分子环境的“小”变化和(ii)与特定原子和原子对定性和定量相关。所描述的协议同样适用于 (a) 任何核间区域, (b) 无论涉及哪种相互作用 (吸引力/排斥) 原子, (c) 在用于计算分子结构和相应波函数的任何理论水平, 和 (d) 平衡或非平衡结构。最后,我们主张从 ED 的角度对化学相互作用的描述进行范式转变,以支持多中心而不是双原子方法来解释核间区域的 ED 分布。© 2018 Wiley Periodicals, Inc.

























 京公网安备 11010802027423号
京公网安备 11010802027423号