当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Assessing the capability of in silico mutation protocols for predicting the finite temperature conformation of amino acids†
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2018-10-03 00:00:00 , DOI: 10.1039/c8cp03826k Rodrigo Ochoa 1, 2, 3, 4, 5 , Miguel A. Soler 6, 7, 8 , Alessandro Laio 6, 7, 8, 9, 10 , Pilar Cossio 1, 2, 3, 4, 5
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2018-10-03 00:00:00 , DOI: 10.1039/c8cp03826k Rodrigo Ochoa 1, 2, 3, 4, 5 , Miguel A. Soler 6, 7, 8 , Alessandro Laio 6, 7, 8, 9, 10 , Pilar Cossio 1, 2, 3, 4, 5
Affiliation
|
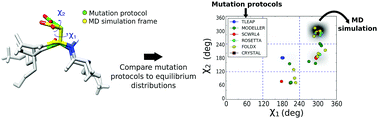
Mutation protocols are a key tool in computational biophysics for modelling unknown side chain conformations. In particular, these protocols are used to generate the starting structures for molecular dynamics simulations. The accuracy of the initial side chain and backbone placement is crucial to obtain a stable and quickly converging simulation. In this work, we assessed the performance of several mutation protocols in predicting the most probable conformer observed in finite temperature molecular dynamics simulations for a set of protein–peptide crystals differing only by single-point mutations in the peptide sequence. Our results show that several programs which predict well the crystal conformations fail to predict the most probable finite temperature configuration. Methods relying on backbone-dependent rotamer libraries have, in general, a better performance, but even the best protocol fails in predicting approximately 30% of the mutations.
中文翻译:

评估计算机突变协议预测氨基酸有限温度构象的能力†
突变协议是计算生物物理学中用于建模未知侧链构象的关键工具。特别是,这些协议用于生成分子动力学模拟的起始结构。初始侧链和主干位置的准确性对于获得稳定且快速收敛的仿真至关重要。在这项工作中,我们评估了几种突变方案在预测一组蛋白质-肽晶体的有限温度分子动力学模拟中观察到的最可能的构象异构体方面的性能,该蛋白质-肽晶体的区别仅在于肽序列中的单点突变。我们的结果表明,几个能很好地预测晶体构象的程序无法预测最可能的有限温度构型。通常,依赖于依赖于主链的旋转异构体文库的方法具有
更新日期:2018-10-03
中文翻译:
评估计算机突变协议预测氨基酸有限温度构象的能力†
突变协议是计算生物物理学中用于建模未知侧链构象的关键工具。特别是,这些协议用于生成分子动力学模拟的起始结构。初始侧链和主干位置的准确性对于获得稳定且快速收敛的仿真至关重要。在这项工作中,我们评估了几种突变方案在预测一组蛋白质-肽晶体的有限温度分子动力学模拟中观察到的最可能的构象异构体方面的性能,该蛋白质-肽晶体的区别仅在于肽序列中的单点突变。我们的结果表明,几个能很好地预测晶体构象的程序无法预测最可能的有限温度构型。通常,依赖于依赖于主链的旋转异构体文库的方法具有

























 京公网安备 11010802027423号
京公网安备 11010802027423号