当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
A quantitative connection of experimental and simulated folding landscapes by vibrational spectroscopy†
Chemical Science ( IF 8.4 ) Pub Date : 2018-10-03 00:00:00 , DOI: 10.1039/c8sc03786h Caitlin M Davis 1, 2 , Laura Zanetti-Polzi 3 , Martin Gruebele 1, 4 , Andrea Amadei 5 , R Brian Dyer 2 , Isabella Daidone 3
Chemical Science ( IF 8.4 ) Pub Date : 2018-10-03 00:00:00 , DOI: 10.1039/c8sc03786h Caitlin M Davis 1, 2 , Laura Zanetti-Polzi 3 , Martin Gruebele 1, 4 , Andrea Amadei 5 , R Brian Dyer 2 , Isabella Daidone 3
Affiliation
|
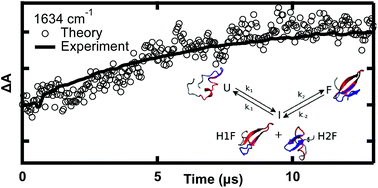
For small molecule reaction kinetics, computed reaction coordinates often mimic experimentally measured observables quite accurately. Although nowadays simulated and measured biomolecule kinetics can be compared on the same time scale, a gap between computed and experimental observables remains. Here we directly compared temperature-jump experiments and molecular dynamics simulations of protein folding dynamics using the same observable: the time-dependent infrared spectrum. We first measured the stability and folding kinetics of the fastest-folding β-protein, the GTT35 WW domain, using its structurally specific infrared spectrum. The relaxation dynamics of the peptide backbone, β-sheets, turn, and random coil were measured independently by probing the amide I′ region at different frequencies. Next, the amide I′ spectra along folding/unfolding molecular dynamics trajectories were simulated by accurate mixed quantum/classical calculations. The simulated time dependence and spectral amplitudes at the exact experimental probe frequencies provided relaxation and folding rates in agreement with experimental observations. The calculations validated by experiment yield direct structural evidence for a rate-limiting reaction step where an intermediate state with either the first or second hairpin is formed. We show how folding switches from a more homogeneous (apparent two-state) process at high temperature to a more heterogeneous process at low temperature, where different parts of the WW domain fold at different rates.
中文翻译:

通过振动光谱定量连接实验和模拟折叠景观†
对于小分子反应动力学,计算出的反应坐标通常非常准确地模拟实验测量的可观察量。尽管现在可以在相同的时间尺度上比较模拟和测量的生物分子动力学,但计算和实验观测值之间仍然存在差距。在这里,我们使用相同的观测值直接比较了温度跳跃实验和蛋白质折叠动力学的分子动力学模拟:时间相关的红外光谱。我们首先使用其结构特异性红外光谱测量了折叠速度最快的 β 蛋白 GTT35 WW 结构域的稳定性和折叠动力学。通过以不同频率探测酰胺 I' 区域,独立测量肽骨架、β-折叠、转角和无规卷曲的弛豫动力学。下一个,通过精确的混合量子/经典计算模拟了沿折叠/展开分子动力学轨迹的酰胺 I' 光谱。在精确的实验探针频率下模拟的时间依赖性和光谱幅度提供了与实验观察一致的弛豫和折叠率。通过实验验证的计算为限速反应步骤提供了直接的结构证据,其中形成了具有第一或第二发夹的中间状态。我们展示了折叠如何从高温下的更均匀(明显的两态)过程转换为低温下更不均匀的过程,其中 WW 域的不同部分以不同的速率折叠。在精确的实验探针频率下模拟的时间依赖性和光谱幅度提供了与实验观察一致的弛豫和折叠率。通过实验验证的计算为限速反应步骤提供了直接的结构证据,其中形成了具有第一或第二发夹的中间状态。我们展示了折叠如何从高温下的更均匀(明显的两态)过程转换为低温下更不均匀的过程,其中 WW 域的不同部分以不同的速率折叠。在精确的实验探针频率下模拟的时间依赖性和光谱幅度提供了与实验观察一致的弛豫和折叠率。通过实验验证的计算为限速反应步骤提供了直接的结构证据,其中形成了具有第一或第二发夹的中间状态。我们展示了折叠如何从高温下的更均匀(明显的两态)过程转换为低温下更不均匀的过程,其中 WW 域的不同部分以不同的速率折叠。通过实验验证的计算为限速反应步骤提供了直接的结构证据,其中形成了具有第一或第二发夹的中间状态。我们展示了折叠如何从高温下的更均匀(明显的两态)过程转换为低温下更不均匀的过程,其中 WW 域的不同部分以不同的速率折叠。通过实验验证的计算为限速反应步骤提供了直接的结构证据,其中形成了具有第一或第二发夹的中间状态。我们展示了折叠如何从高温下的更均匀(明显的两态)过程转换为低温下更不均匀的过程,其中 WW 域的不同部分以不同的速率折叠。
更新日期:2018-10-03
中文翻译:
通过振动光谱定量连接实验和模拟折叠景观†
对于小分子反应动力学,计算出的反应坐标通常非常准确地模拟实验测量的可观察量。尽管现在可以在相同的时间尺度上比较模拟和测量的生物分子动力学,但计算和实验观测值之间仍然存在差距。在这里,我们使用相同的观测值直接比较了温度跳跃实验和蛋白质折叠动力学的分子动力学模拟:时间相关的红外光谱。我们首先使用其结构特异性红外光谱测量了折叠速度最快的 β 蛋白 GTT35 WW 结构域的稳定性和折叠动力学。通过以不同频率探测酰胺 I' 区域,独立测量肽骨架、β-折叠、转角和无规卷曲的弛豫动力学。下一个,通过精确的混合量子/经典计算模拟了沿折叠/展开分子动力学轨迹的酰胺 I' 光谱。在精确的实验探针频率下模拟的时间依赖性和光谱幅度提供了与实验观察一致的弛豫和折叠率。通过实验验证的计算为限速反应步骤提供了直接的结构证据,其中形成了具有第一或第二发夹的中间状态。我们展示了折叠如何从高温下的更均匀(明显的两态)过程转换为低温下更不均匀的过程,其中 WW 域的不同部分以不同的速率折叠。在精确的实验探针频率下模拟的时间依赖性和光谱幅度提供了与实验观察一致的弛豫和折叠率。通过实验验证的计算为限速反应步骤提供了直接的结构证据,其中形成了具有第一或第二发夹的中间状态。我们展示了折叠如何从高温下的更均匀(明显的两态)过程转换为低温下更不均匀的过程,其中 WW 域的不同部分以不同的速率折叠。在精确的实验探针频率下模拟的时间依赖性和光谱幅度提供了与实验观察一致的弛豫和折叠率。通过实验验证的计算为限速反应步骤提供了直接的结构证据,其中形成了具有第一或第二发夹的中间状态。我们展示了折叠如何从高温下的更均匀(明显的两态)过程转换为低温下更不均匀的过程,其中 WW 域的不同部分以不同的速率折叠。通过实验验证的计算为限速反应步骤提供了直接的结构证据,其中形成了具有第一或第二发夹的中间状态。我们展示了折叠如何从高温下的更均匀(明显的两态)过程转换为低温下更不均匀的过程,其中 WW 域的不同部分以不同的速率折叠。通过实验验证的计算为限速反应步骤提供了直接的结构证据,其中形成了具有第一或第二发夹的中间状态。我们展示了折叠如何从高温下的更均匀(明显的两态)过程转换为低温下更不均匀的过程,其中 WW 域的不同部分以不同的速率折叠。

























 京公网安备 11010802027423号
京公网安备 11010802027423号