当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Electron Propagator Methods for Vertical Electron Detachment Energies of Anions: Benchmarks and Case Studies
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-09-25 00:00:00 , DOI: 10.1021/acs.jctc.8b00736 Manuel Díaz-Tinoco 1 , H. H. Corzo 1 , J. V. Ortiz 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-09-25 00:00:00 , DOI: 10.1021/acs.jctc.8b00736 Manuel Díaz-Tinoco 1 , H. H. Corzo 1 , J. V. Ortiz 1
Affiliation
|
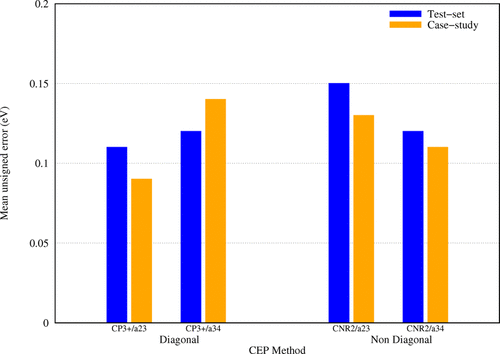
Ab initio electron propagator methods are efficient and accurate means of calculating vertical electron detachment energies of closed-shell, molecular anions with nuclei from the first three periods. Basis set extrapolations enable definitive comparisons between electron propagator results and benchmarks defined by total energy differences obtained with coupled-cluster, single, double, plus perturbative triple substitution theory. The best compromises of accuracy and efficiency are provided by the renormalized, partial third-order, diagonal (P3+) self-energy and by the nondiagonal, renormalized, second-order (NR2) approximation. The outer-valence Green function, the two-particle-one-hole Tamm–Dancoff approximation, the third-order algebraic diagrammatic construction, and the renormalized third-order methods also are examined. A detailed analysis of errors for small anions is performed. Case studies include F–(H2O) and Cl–(H2O) complexes, C5H5–, two P2N3– pentagonal rings, and a superhalide, Al(BO2)4–, whose electron detachment energy is more than double those of the halide anions. These applications illustrate the versatility of electron propagator methods, their utility for interpreting negative-ion photoelectron spectra, and their promise in the discovery of unusual properties and patterns of chemical bonding. Composite methods, which combine basis set effects calculated at the relatively efficient diagonal, second-order level and higher correlation effects calculated with small basis sets, provide excellent estimates of basis set-extrapolated P3+ or NR2 results and facilitate applications to large molecules. In the P3+ and NR2 methods, a judicious choice of low-order couplings between hole operators that correspond to the assumptions of Koopmans’s theorem and operators that describe final-state relaxation and polarization and initial-state correlation leads to predictive accuracy, computational efficiency, and interpretive lucidity.
中文翻译:

阴离子的垂直电子脱附能的电子传播方法:基准和案例研究
从头算电子传播器方法是计算前三个周期中带有核的闭壳分子阴离子的垂直电子离解能的高效且准确的方法。通过基集外推法,可以在电子传播器结果与基准之间进行确定的比较,这些基准是由耦合簇,单,双和微扰三取代理论获得的总能量差定义的。重新归一化的部分三阶对角(P3 +)自能和非对角,重新归一化的二阶(NR2)近似提供了准确性和效率的最佳折衷。还检验了外价格林函数,两粒子一孔的Tamm–Dancoff逼近,三阶代数图解结构以及重新归一化的三阶方法。对小阴离子的误差进行了详细分析。案例研究包括F-(H 2 O)和Cl -(H 2 O)配合物,C 5 ħ 5 - ,两个P 2 Ñ 3 -五边形环和一个superhalide,铝(BO 2)4 -,其电子离解能是卤化物阴离子的两倍以上。这些应用说明了电子传播方法的多功能性,其用于解释负离子光电子能谱的效用以及它们在发现异常性质和化学键合模式方面的前景。组合方法结合了在相对有效的对角线,二阶水平上计算出的基集效果和使用小基集计算出的较高相关性效果,可提供对基集外推的P3 +或NR2结果的出色估计,并便于应用于大分子。在P3 +和NR2方法中,
更新日期:2018-09-25
中文翻译:
阴离子的垂直电子脱附能的电子传播方法:基准和案例研究
从头算电子传播器方法是计算前三个周期中带有核的闭壳分子阴离子的垂直电子离解能的高效且准确的方法。通过基集外推法,可以在电子传播器结果与基准之间进行确定的比较,这些基准是由耦合簇,单,双和微扰三取代理论获得的总能量差定义的。重新归一化的部分三阶对角(P3 +)自能和非对角,重新归一化的二阶(NR2)近似提供了准确性和效率的最佳折衷。还检验了外价格林函数,两粒子一孔的Tamm–Dancoff逼近,三阶代数图解结构以及重新归一化的三阶方法。对小阴离子的误差进行了详细分析。案例研究包括F-(H 2 O)和Cl -(H 2 O)配合物,C 5 ħ 5 - ,两个P 2 Ñ 3 -五边形环和一个superhalide,铝(BO 2)4 -,其电子离解能是卤化物阴离子的两倍以上。这些应用说明了电子传播方法的多功能性,其用于解释负离子光电子能谱的效用以及它们在发现异常性质和化学键合模式方面的前景。组合方法结合了在相对有效的对角线,二阶水平上计算出的基集效果和使用小基集计算出的较高相关性效果,可提供对基集外推的P3 +或NR2结果的出色估计,并便于应用于大分子。在P3 +和NR2方法中,


























 京公网安备 11010802027423号
京公网安备 11010802027423号