当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Protein Conformational Transitions from All-Atom Adaptively Biased Path Optimization
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-09-17 00:00:00 , DOI: 10.1021/acs.jctc.8b00147 Heng Wu 1 , Carol Beth Post 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-09-17 00:00:00 , DOI: 10.1021/acs.jctc.8b00147 Heng Wu 1 , Carol Beth Post 1
Affiliation
|
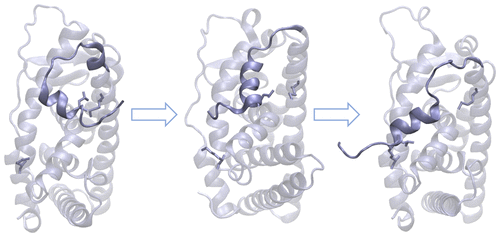
Simulation methods are valuable for elucidating protein conformational transitions between functionally diverse states given that transition pathways are difficult to capture experimentally. Nonetheless, specific computational algorithms are required because of the high free energy barriers between these different protein conformational states. Adaptively biased path optimization (ABPO) is an unrestrained, transition-path optimization method that works in a reduced-variable space to construct an adaptive biasing potential to aid convergence. ABPO was previously applied using a coarse-grained Go̅-model to study conformational activation of Lyn, a Src family tyrosine kinase. How effectively ABPO can be applied at the higher resolution of an all-atom model to explore protein conformational transitions is not yet known. Here, we report the all-atom conformational transition paths of three protein systems constructed using the ABPO methodology. Two systems, triose phosphate isomerase and dihydrofolate reductase, undergo local flipping of a short loop that promotes ligand binding. The third system, estrogen receptor α ligand binding domain, has a helix that adopts different conformations when the protein is bound to an agonist or an antagonist. For each protein, distance-based or torsion-angle reduced variables were identified from unbiased trajectories. ABPO was computed in this reduced variable space to obtain the transition path between the two states. The all-atom ABPO is shown to successfully converge an optimal transition path for each of the three systems.
中文翻译:

从全原子自适应偏置路径优化的蛋白质构象转变。
考虑到难以通过实验捕获过渡途径,模拟方法对于阐明功能多样化状态之间的蛋白质构象过渡非常有价值。但是,由于这些不同蛋白质构象状态之间的自由能垒很高,因此需要特定的计算算法。自适应偏置路径优化(ABPO)是一种无约束的过渡路径优化方法,该方法在减小的变量空间中工作,以构建自适应偏置电势以帮助收敛。以前使用粗粒Go̅模型应用ABPO来研究Lyn的构象活化,Lyn是Src家族的酪氨酸激酶。尚不知道如何在全原子模型的高分辨率下有效应用ABPO来探索蛋白质构象转变。这里,我们报告了使用ABPO方法构建的三个蛋白质系统的全原子构象过渡路径。磷酸三糖异构酶和二氢叶酸还原酶这两个系统经历了促进配体结合的短环的局部翻转。第三个系统是雌激素受体α配体结合域,其螺旋结构在蛋白质与激动剂或拮抗剂结合时会采用不同的构象。对于每种蛋白质,从无偏轨中识别出基于距离或扭转角减小的变量。在这个减少的变量空间中计算ABPO,以获得两个状态之间的过渡路径。已显示全原子ABPO已成功收敛了三个系统中每个系统的最佳过渡路径。磷酸丙糖异构酶和二氢叶酸还原酶经历短环的局部翻转,从而促进配体结合。第三个系统是雌激素受体α配体结合域,其螺旋结构在蛋白质与激动剂或拮抗剂结合时会采用不同的构象。对于每种蛋白质,从无偏轨中识别出基于距离或扭转角减小的变量。在这个减少的变量空间中计算ABPO,以获得两个状态之间的过渡路径。已显示全原子ABPO已成功收敛了三个系统中每个系统的最佳过渡路径。磷酸丙糖异构酶和二氢叶酸还原酶经历短环的局部翻转,从而促进配体结合。第三个系统是雌激素受体α配体结合域,其螺旋结构在蛋白质与激动剂或拮抗剂结合时会采用不同的构象。对于每种蛋白质,从无偏轨中识别出基于距离或扭转角减小的变量。在这个减少的变量空间中计算ABPO,以获得两个状态之间的过渡路径。已显示全原子ABPO已成功收敛了三个系统中每个系统的最佳过渡路径。当蛋白质与激动剂或拮抗剂结合时,其螺旋结构会采用不同的构象。对于每种蛋白质,从无偏轨中识别出基于距离或扭转角减小的变量。在这个减少的变量空间中计算ABPO,以获得两个状态之间的过渡路径。已显示全原子ABPO已成功收敛了三个系统中每个系统的最佳过渡路径。当蛋白质与激动剂或拮抗剂结合时,其螺旋结构会采用不同的构象。对于每种蛋白质,从无偏轨中识别出基于距离或扭转角减小的变量。在这个减少的变量空间中计算ABPO,以获得两个状态之间的过渡路径。已显示全原子ABPO已成功收敛了三个系统中每个系统的最佳过渡路径。
更新日期:2018-09-17
中文翻译:
从全原子自适应偏置路径优化的蛋白质构象转变。
考虑到难以通过实验捕获过渡途径,模拟方法对于阐明功能多样化状态之间的蛋白质构象过渡非常有价值。但是,由于这些不同蛋白质构象状态之间的自由能垒很高,因此需要特定的计算算法。自适应偏置路径优化(ABPO)是一种无约束的过渡路径优化方法,该方法在减小的变量空间中工作,以构建自适应偏置电势以帮助收敛。以前使用粗粒Go̅模型应用ABPO来研究Lyn的构象活化,Lyn是Src家族的酪氨酸激酶。尚不知道如何在全原子模型的高分辨率下有效应用ABPO来探索蛋白质构象转变。这里,我们报告了使用ABPO方法构建的三个蛋白质系统的全原子构象过渡路径。磷酸三糖异构酶和二氢叶酸还原酶这两个系统经历了促进配体结合的短环的局部翻转。第三个系统是雌激素受体α配体结合域,其螺旋结构在蛋白质与激动剂或拮抗剂结合时会采用不同的构象。对于每种蛋白质,从无偏轨中识别出基于距离或扭转角减小的变量。在这个减少的变量空间中计算ABPO,以获得两个状态之间的过渡路径。已显示全原子ABPO已成功收敛了三个系统中每个系统的最佳过渡路径。磷酸丙糖异构酶和二氢叶酸还原酶经历短环的局部翻转,从而促进配体结合。第三个系统是雌激素受体α配体结合域,其螺旋结构在蛋白质与激动剂或拮抗剂结合时会采用不同的构象。对于每种蛋白质,从无偏轨中识别出基于距离或扭转角减小的变量。在这个减少的变量空间中计算ABPO,以获得两个状态之间的过渡路径。已显示全原子ABPO已成功收敛了三个系统中每个系统的最佳过渡路径。磷酸丙糖异构酶和二氢叶酸还原酶经历短环的局部翻转,从而促进配体结合。第三个系统是雌激素受体α配体结合域,其螺旋结构在蛋白质与激动剂或拮抗剂结合时会采用不同的构象。对于每种蛋白质,从无偏轨中识别出基于距离或扭转角减小的变量。在这个减少的变量空间中计算ABPO,以获得两个状态之间的过渡路径。已显示全原子ABPO已成功收敛了三个系统中每个系统的最佳过渡路径。当蛋白质与激动剂或拮抗剂结合时,其螺旋结构会采用不同的构象。对于每种蛋白质,从无偏轨中识别出基于距离或扭转角减小的变量。在这个减少的变量空间中计算ABPO,以获得两个状态之间的过渡路径。已显示全原子ABPO已成功收敛了三个系统中每个系统的最佳过渡路径。当蛋白质与激动剂或拮抗剂结合时,其螺旋结构会采用不同的构象。对于每种蛋白质,从无偏轨中识别出基于距离或扭转角减小的变量。在这个减少的变量空间中计算ABPO,以获得两个状态之间的过渡路径。已显示全原子ABPO已成功收敛了三个系统中每个系统的最佳过渡路径。

























 京公网安备 11010802027423号
京公网安备 11010802027423号