当前位置:
X-MOL 学术
›
Small Methods
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Property–Activity Relations for Methane Activation by Dual‐Metal Cu–Oxo Trimers in ZSM‐5 Zeolite
Small Methods ( IF 12.4 ) Pub Date : 2018-08-24 , DOI: 10.1002/smtd.201800266 Chong Liu 1 , Guanna Li 1, 2 , Evgeny A. Pidko 1, 3
Small Methods ( IF 12.4 ) Pub Date : 2018-08-24 , DOI: 10.1002/smtd.201800266 Chong Liu 1 , Guanna Li 1, 2 , Evgeny A. Pidko 1, 3
Affiliation
|
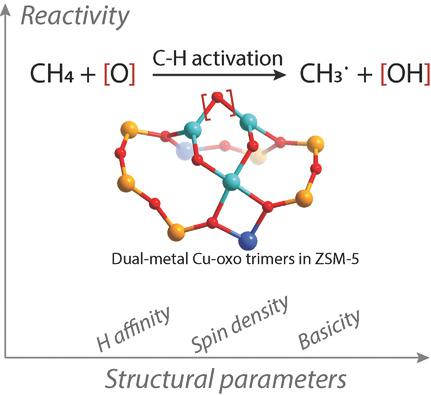
The identification of correlations between the experimentally or computationally measurable parameters of a catalytic system and its reactivity is one of the key steps toward the realization of a catalysis by design strategy. Here, periodic density functional theory calculations to establish such correlations for perspective catalysts for selective methane oxidation based on dual‐metal cation‐exchanged zeolites are employed. A representative trimeric metal–oxo active site is considered as a model reactive center. Computations reveal that the activation barrier for the homolytic CH bond cleavage in methane correlates well with the thermodynamic stability of the resulting CH3·⋯HO intermediate. The stability of the HO part approximated by the hydrogen affinity of the active site correlates with the activity trend, but deviations are observed due to inability of this descriptor to account for the stabilization of the CH3∙ moiety. Such fundamental characteristics as the atomic spin density and the basicity of reactive oxygen sites cannot be directly correlated with the catalyst reactivity, implying the complexity in property–reactivity relationship for methane activation. Calculations suggest that the initial screening of the potent zeolite‐based catalyst for methane activation can be established based on the analysis of both the thermodynamics and perturbation of base molecular adsorption probes such as pyrrole.
中文翻译:

ZSM-5沸石中双金属铜-氧三聚体活化甲烷的特性-活性关系
识别催化系统的实验或计算可测量参数与其反应性之间的相关性是通过设计策略实现催化的关键步骤之一。在这里,采用周期性密度泛函理论计算来建立基于双金属阳离子交换沸石的选择性甲烷氧化透视催化剂的相关性。代表性的三聚体金属-羰基活性位点被认为是模型反应中心。计算表明,甲烷中均相CH键裂解的活化障碍与所得CH 3的热力学稳定性密切相关。·⋯HO中间体。通过活性位点的氢亲和力估算出的HO部分的稳定性与活性趋势相关,但是由于该描述符无法解释CH 3 ∙部分的稳定性,因此观察到偏差。诸如原子自旋密度和活性氧位的碱度等基本特征不能与催化剂的反应性直接相关,这意味着甲烷活化的性质-反应性关系很复杂。计算表明,可以基于对基本分子吸附探针(如吡咯)的热力学和扰动分析,对有效的基于沸石的甲烷活化催化剂进行初步筛选。
更新日期:2018-08-24
中文翻译:
ZSM-5沸石中双金属铜-氧三聚体活化甲烷的特性-活性关系
识别催化系统的实验或计算可测量参数与其反应性之间的相关性是通过设计策略实现催化的关键步骤之一。在这里,采用周期性密度泛函理论计算来建立基于双金属阳离子交换沸石的选择性甲烷氧化透视催化剂的相关性。代表性的三聚体金属-羰基活性位点被认为是模型反应中心。计算表明,甲烷中均相CH键裂解的活化障碍与所得CH 3的热力学稳定性密切相关。·⋯HO中间体。通过活性位点的氢亲和力估算出的HO部分的稳定性与活性趋势相关,但是由于该描述符无法解释CH 3 ∙部分的稳定性,因此观察到偏差。诸如原子自旋密度和活性氧位的碱度等基本特征不能与催化剂的反应性直接相关,这意味着甲烷活化的性质-反应性关系很复杂。计算表明,可以基于对基本分子吸附探针(如吡咯)的热力学和扰动分析,对有效的基于沸石的甲烷活化催化剂进行初步筛选。


























 京公网安备 11010802027423号
京公网安备 11010802027423号